Article Text

Statistics from Altmetric.com
Advances in genetics and molecular biology have led to a better understanding of the control of central nervous system (CNS) development. It is possible to classify CNS abnormalities according to the developmental stages at which they occur, as is shown below. The careful assessment of patients with these abnormalities is important in order to provide an accurate prognosis and genetic counselling.
NORMAL DEVELOPMENT OF THE CNS
Before we review the various abnormalities that can affect the CNS, a brief overview of the normal development of the CNS is appropriate.
-
Induction—After development of the three cell layers of the early embryo (ectoderm, mesoderm, and endoderm), the underlying mesoderm (the “inducer”) sends signals to a region of the ectoderm (the “induced tissue”), instructing it to develop into neural tissue.
-
Neural tube formation—The neural ectoderm folds to form a tube, which runs for most of the length of the embryo.
-
Regionalisation and specification—Specification of different regions and individual cells within the neural tube occurs in both the rostral/caudal and dorsal/ventral axis. The three basic regions of the CNS (forebrain, midbrain, and hindbrain) develop at the rostral end of the tube, with the spinal cord more caudally. Within the developing spinal cord specification of the different populations of neural precursors (neural crest, sensory neurones, interneurones, glial cells, and motor neurones) is observed in progressively more ventral locations. This process results from the interaction between genes whose expression defines individual territories or cell types, and diffusible signalling molecules (such as sonic hedgehog) secreted by adjacent areas of the embryo.
-
Proliferation and migration—The most dorsal cells of the tube (the neural crest) migrate away to form much of the peripheral nervous system. Cell proliferation within the tube leads to thickening of the wall and many different cell types move to their correct locations. The development of the forebrain cortex provides a good example. An area called the germinal matrix adjacent to the lumen of the neural tube (the future ventricular system) contains neural stem cells that are precursors of the neurones and of the two glial cell types, oligodendrocytes and astrocytes. Neuronal precursor cells migrate, often along specialised cells called radial glial cells, to their final and particular locations in one of the six layers of the cerebral cortex.
-
Connection and selection—Once each cell is specified according to type and is in an appropriate location, axon outgrowth and synapse formation occurs. The mechanisms that control these connections are complex and incompletely understood. Cells failing to establish the correct connections undergo programmed cell death (apoptosis) as a result of a failure to obtain survival factors produced by the target cells.
DISORDERS OF NEURAL TUBE FORMATION
The neural tube usually fuses 18–26 days after ovulation. Failure of closure may lead to anencephaly, encephalocele, spina bifida or spina bifida occulta. Liveborn anencephalic babies usually die in hours or days.
Epidemiology
Neural tube defects (NTDs) are among the most common congenital abnormalities but prevalence varies between countries and races. The prevalence of NTDs in England and Wales fell from the 1970s onwards to just under 0.8/1000 total births by 1994. Some of the decline was due to antenatal diagnosis but some is unexplained. In the UK anencephaly and spina bifida are of approximately equal prevalence and together make up 95% of all NTDs.
Aetiology
Most NTDs result from a complex interaction between several genes and poorly understood environmental factors.
Genetic factors
NTDs occur in many syndromes and chromosome disorders. However, an NTD may be the only anomaly in a member of a family in which case the relatives have an increased risk for all types of NTD.
Environmental factors
Periconceptual multiple vitamin supplements containing folic acid reduce the incidence of neural tube defects. In England it is recommended that women planning pregnancy take 400 μg of folic acid daily before conception and during the first 12 weeks of the pregnancy. Some drugs taken during the pregnancy may increase the risk. These include sodium valproate and folic acid antagonists such as trimethoprim, triamterene, carbamazepine, phenytoin, phenobarbitone, and primidone.
Prenatal diagnosis
α Fetoprotein (AFP) concentrations in maternal serum provide a means of screening for open NTDs which allow AFP from the fetal liver to leak into amniotic fluid and then maternal blood, best detected at 16–18 weeks of pregnancy. Ultrasonography is recommended for at-risk women (positive serum AFP screening, previously affected child, taking drugs associated with NTDs in the fetus). Ultrasound can detect anencephaly from 12 weeks gestation and spina bifida from 16–20 weeks. However, spina bifida may be missed, particularly in the L5–S2 region. Amniocentesis to detect raised amniotic fluid AFP has been largely superseded by detailed ultrasound imaging.
Spinal dysraphism
Spina bifida cystica
This is a cystic lesion which in 80–90% is a myelomeningocele in which the spinal cord is a component of the cyst wall. It is lumbosacral in about 80% of cases. There is usually a mixture of upper and lower motor neurone signs depending on the level and there is always disturbance of bladder and bowel function. Surviving infants require complex orthopaedic and urological support, including surgery. About 5% of cases of spina bifida cystica are meningoceles in which there is no neural tissue in the cyst wall, there is no associated hydrocephalus, and neurological examination may be normal.
Hydrocephalus complicates most cases of lumbosacral meningomyelocele. Ultrasound shows hydrocephalus in about 90% of cases at birth. Usually it is associated with the Chiari II malformation, which is present in about 70% of cases of meningomyelocele and consists of downward protrusion of the medulla below the foramen magnum to overlap the spinal cord. Distortion of the medulla and midbrain can cause lower cranial nerve palsies and central apnoea (which may be misdiagnosed as epilepsy).
Treatment of infants with meningomyeloceles became possible with the development of ventriculo-atrial and ventriculo-peritoneal shunts. The early active management involves closing the defect and inserting a shunt. Active intervention in all cases was questioned in the 1970s and a more selective approach was advocated. However, selective surgical management is not universally practised and this remains a controversial area.
Occult spinal dysraphism
The term spina bifida occulta is often applied to a defect of the posterior arch of one or more lumbar or sacral vertebrae (usually L5 and S1). It is found incidentally by x ray in 25% of hospitalised children and may be regarded as a normal variant. However, it must not be assumed that spina bifida occulta is always benign. If examination of the skin over the spine reveals a naevus, hairy patch, sinus or subcutaneous mass, magnetic resonance imaging of the spinal cord is probably indicated, particularly if there are associated neurological abnormalities of sphincter or limb control. Several different abnormalities may be found, such as an open sinus tract which could cause recurrent meningitis, a lipoma attached to a low lying spinal cord, or diastematomyelia which is a sagittal cleft dividing the spinal cord into two halves often with a bony or cartilaginous spur transfixing the cord. If an abnormality involving the cord or nerve roots is found there is a case for neurosurgical intervention, but the indications for this are controversial.
Syringomyelia
This is a tubular cavitation of the spinal cord which tends to be in the cervical region but may involve the whole cord. It rarely becomes symptomatic in children. Shunting of the cavity is sometimes performed and posterior fossa exploration may be undertaken. It is often associated with the Chiari I malformation in which there is downward displacement of the lower cerebellum, including the tonsils (fig 1).

Chiari I malformation. Sagittal T1 weighted magnetic resonance image showing an enlarged cerebellar tonsil which extends below the level of the foramen magnum (arrow). The corpus callosum is normal with the cingulate gyrus just above it (compare to figure 5). This 12 year old boy presented with headaches. The Chiari I malformation can be associated with hydrocephalus and syringomyelia, which he did not have.
DISORDERS OF REGIONALISATION
Abnormal development of the anterior portion of the neural tube (the mediobasal prosencephalon) and associated structures caused by disturbances in ventral induction (described above) may cause abnormalities of the brain and face. The most severe is holoprosencephaly in which there is failure of the prosencephalon to separate into two cerebral hemispheres. The mildest is olfactory aplasia without other cerebral malformations. The severity of associated facial abnormalities often parallels those in the brain. In the most severe facial abnormality there is anophthalmia and absence of the nose. However, there may be just mild hypotelorism (closely set eyes), a single central incisor tooth or the face may be normal.
DISORDERS OF CORTICAL DEVELOPMENT
Disorders of proliferation and differentiation
Microcephaly
This is an abnormally small head circumference (< 0.4th centile on occipito-frontal head circumference charts), which is disproportionately small in relation to the rest of the body. The usual implication of this finding is that brain growth is not normal. However, if a small head circumference is detected in the neonatal period it is prudent to perform an x ray of the skull to look for evidence of early closure of all the cranial sutures (total craniosynostosis).
Genetic causes
There are familial cases where the neurological problems are relatively mild. However microcephaly is usually associated with significant abnormalities such as pyramidal tract signs and profound learning difficulties. It is part of more than 450 syndromes listed in the Oxford Dysmorphology Database.
Non-genetic causes
These include ionising radiation in the first two trimesters of pregnancy, intrauterine infections, drugs and other chemicals, circulatory disturbance, and perinatal hypoxic-ischaemic events. When there is a significant perinatal insult to the brain the head circumference may be normal at birth with subsequent failure of growth in the first few months of life. Equally in some types of genetic microcephaly the head size falls off as late as 32–34 weeks of gestation or even after birth, so prenatal diagnosis by ultrasound may be difficult.
Megalencephaly
Megalencephaly is increased size of the brain itself. Large heads can run in normal families but inherited megalencephaly can be associated with significant learning difficulties, neurological abnormalities, and seizures. Hemimegalencephaly is unilateral enlargement of one side of the brain, sometimes the hemisphere only. Associated neurological problems can be severe—intractable seizures, developmental delay, and sometimes hemiparesis.
Disorders of migration
Migrating neurones may fail to reach their intended destination in the cerebral cortex. The abnormalities may be focal or diffuse. If neurones fail to leave the ventricular zone, periventricular heterotopias result. If they fail to complete their migration in the cortex this causes lissencephaly. If only a subpopulation of neurones are affected and others complete migration this causes nodular or band heterotopias.
Agyria-pachygyria (lissencephaly)
There may be complete absence of gyri, in which case the terms agyria or lissencephaly (Greek: “smooth brain”) are used. Pachygyria describes a reduced number of broadened and flat gyri with less folding of the cortex than normal. There may be varying degrees of agyria/pachygyria in the same brain (fig 2).

Type I lissencephaly. Coronal T1 weighted magnetic resonance image. Although the brain can be completely smooth in lissencephaly these patients may have areas of both agyria and pachygyria. This scan illustrates the point. The parietal lobe (arrow 1) shows large abnormal gyri (“pachygyria”). In the left temporal lobe there are more normal gyri (arrow 2). In this boy the occipital lobes (not seen) showed complete agyria. He presented in the first few months of life with developmental delay and infantile spasms; the scan was performed at 8 months of age.
Type I lissencephaly
Here the brain is small with only the primary and sometimes a few secondary gyri. The cortex is thick with the white matter forming a thin rim along the ventricles. Infants with type I lissencephaly may be divided into two groups. The minority have the dysmorphic features of the Miller-Dieker syndrome associated with deletions of 17p13.3, a region which includes the LIS1 gene. The majority have the isolated lissencephaly sequence (ILS) and have no dysmorphic features. This is a heterogeneous group. More than 40% have a deletion of, or mutations within, the LIS1 gene. Mutations in a second gene on the X chromosome, doublecortin (DCX), have also been shown to cause lissencephaly.
Type II lissencephaly or Walker-Warburg syndrome
This is also called “cobblestone lissencephaly” and is a different malformation from type I lissencephaly. The smooth cortex has a granular surface and is covered with meninges that are thickened as a result of mesenchymal proliferation. The clinical features include both nervous system and muscle abnormalities. The infants are very abnormal at birth. They have abnormal eyes with retinal dysplasia, microphthalmia, and anomalies of the anterior segment. There may be hydrocephalus or sometimes microcephaly. Usually there is necrosis of fibres in all muscles, similar to that seen in severe muscular dystrophy, and serum creatine kinase is raised.
Apart from the Walker-Warburg syndrome there are other disorders of muscle, eye, and brain. Recent work suggests that mutations within the dystrophin complex underlie these disorders.
Heterotopias
Periventricular heterotopias are abnormal collections of neurones in the subependymal region. They may be part of a complex malformation syndrome or they may be isolated. They may be clinically silent or associated with seizures and developmental problems. Subcortical heterotopias can be divided into two groups. Nodular heterotopias of grey matter are found in association with other migration disorders and may be the cause of partial seizures. Subcortical laminar heterotopias are also known as band heterotopias or “double cortex”.
Polymicrogyria (microgyria)
This developmental disturbance may occur after the fifth month of pregnancy. The causes are poorly understood but may be genetic, infective or hypoxic (perhaps associated with poor cerebral perfusion). The clinical manifestations depend on the location and extent of the abnormalities. There is a bilateral perisylvian syndrome (or anterior operculum syndrome) in which bilateral opercular abnormalities are seen on magnetic resonance imaging, some of which have the appearance of polymicrogyria (fig 3). These patients have a pseudobulbar palsy with dysarthria, loss of voluntary control of the face and tongue leading to drooling and difficulty feeding. Familial occurrence has been reported.

Bilateral opercular polymicrogyria. Coronal T1 weighted magnetic resonance image. These bilateral abnormalities (arrows) are relatively subtle and were not identified on a computed tomographic examination. In the first year of life this boy developed right sided seizures and development was delayed, particularly in language. Later he had poor tongue and pharyngeal coordination and a right hemiplegia. Despite the lateralisation of some of his neurological signs, the scan abnormalities were bilateral.
Disorders of cortical organisation
Some patients have cortical microdysgenesis—microscopic abnormalities of cortical arrangement that have been described in the brains of patients with epilepsy, autism, schizophrenia, and the fetal alcohol syndrome. The extent to which these findings explain abnormal brain function is an area of active research. Other patients have areas of focal cortical dysplasia which are large enough to be seen on computed tomographic or magnetic resonance imaging scans (fig 4). These dysplasias are a cause of early onset seizures that may be focal or generalised. Resection of cortical dysplasias may improve seizure control.
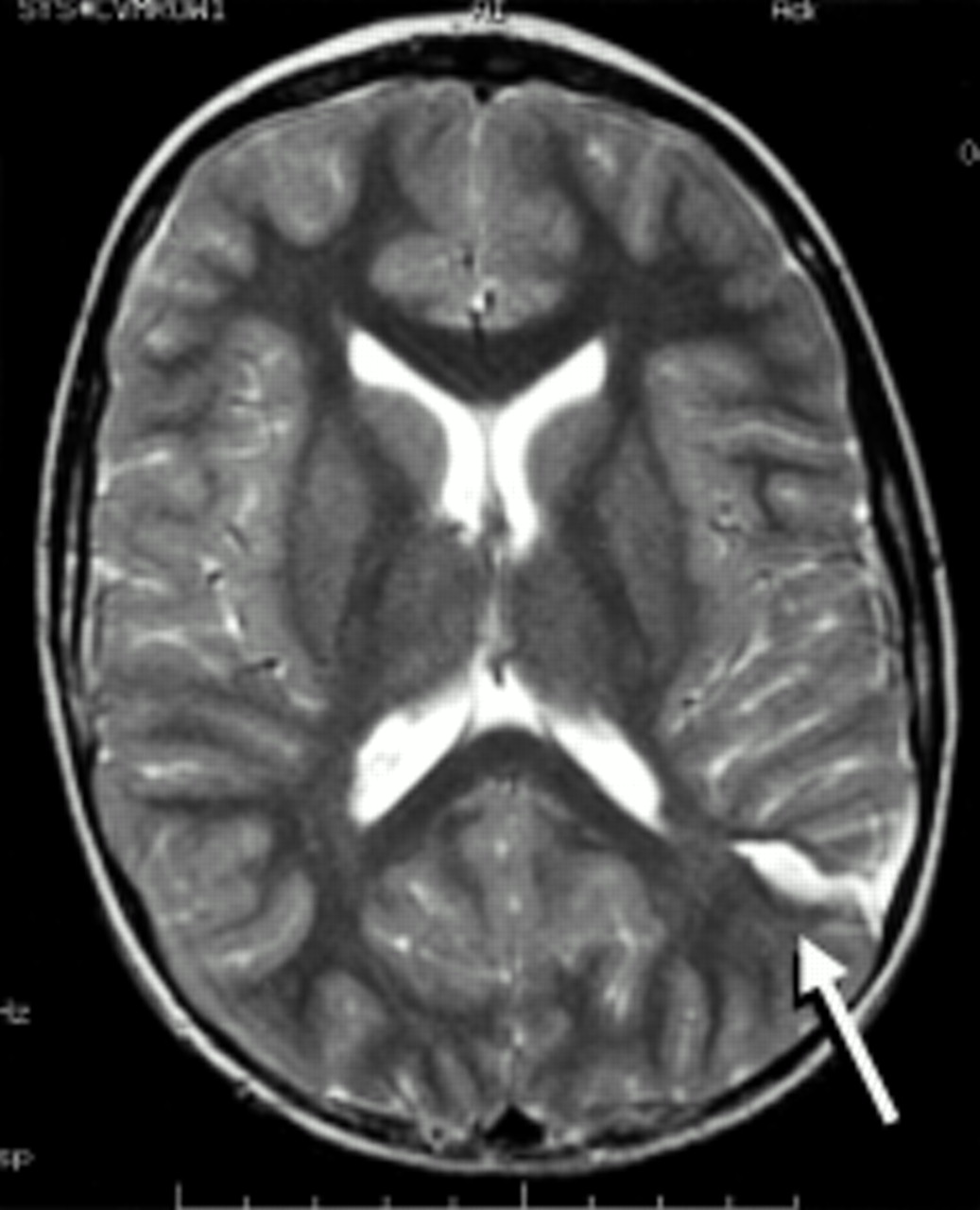
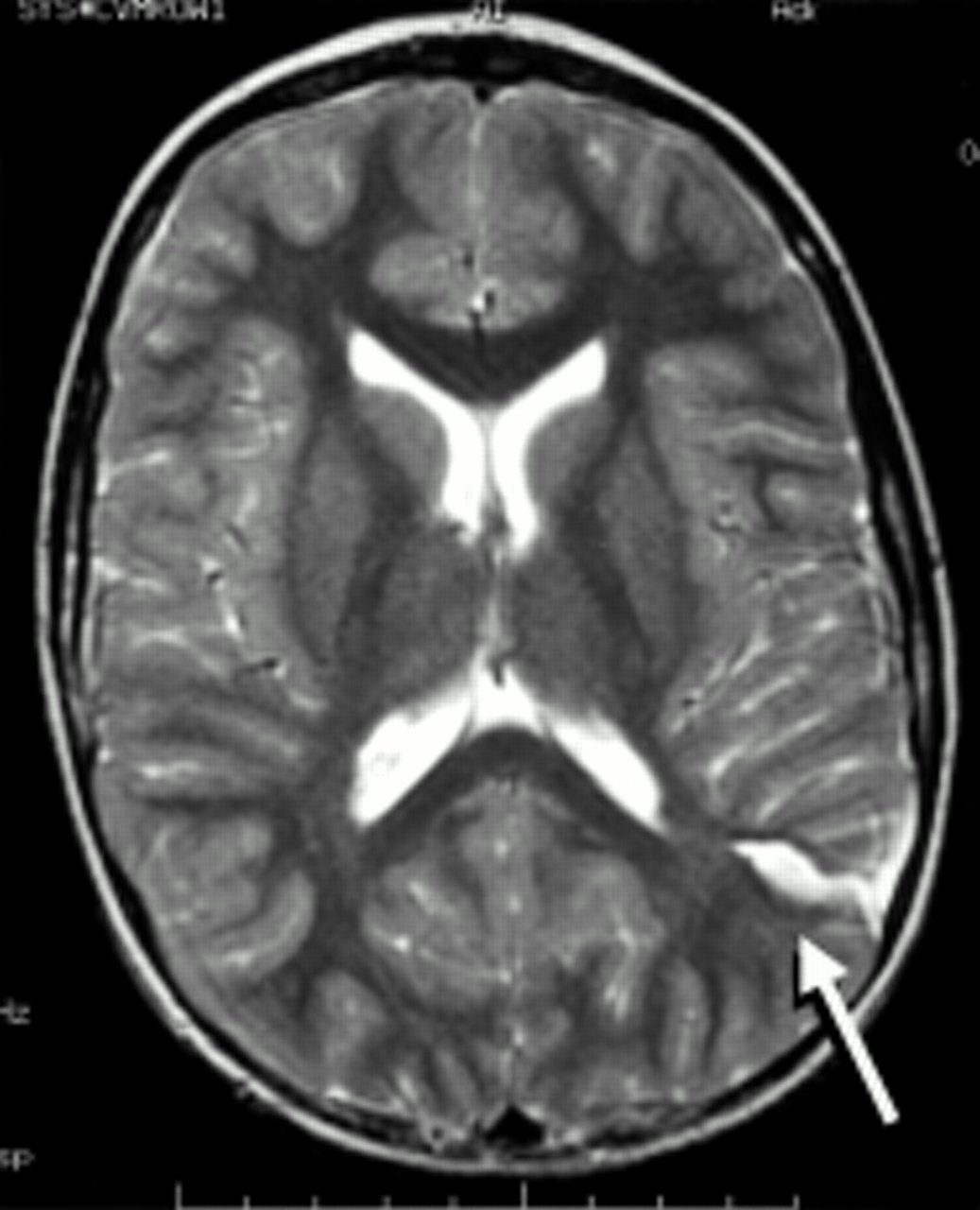
Congenital cortical malformation. Axial T2 weighted magnetic resonance image. There is a cleft in the posterior parietal cortex (arrow) with associated gyral abnormalities more anteriorly. Such lesions are difficult to classify. The term schizencephaly is often used when there is a cleft that extends through the cerebral hemisphere, connecting the ventricle to the subarachnoid space; these clefts tend to be located near the pre- and postcentral gyri. In other cases the malformation may be a localised area of polymicrogyria and the term cortical dysplasia may be used to describe the lesion. This 7 year old girl suffered with partial seizures during which her right arm and hand would go into spasm and then jerk. Between attacks she had slight weakness of the right hand and difficulty performing fine movements.
COMBINED AND OVERLAPPING CEREBRAL MALFORMATIONS
There are distinct abnormalities that represent an overlap between different classes of malformation. This is not surprising—the teratogenic periods are so closely spaced that overlaps are likely if there is an environmental cause. Also in genetically determined syndromes more than one developmental process may be affected.
Agenesis of the corpus callosum
This can be present without symptoms so the true prevalence is not known. Estimated prevalence has varied from 0.05–70 per 10 000 in the general population, increasing to 230 per 10 000 in children with developmental disabilities.
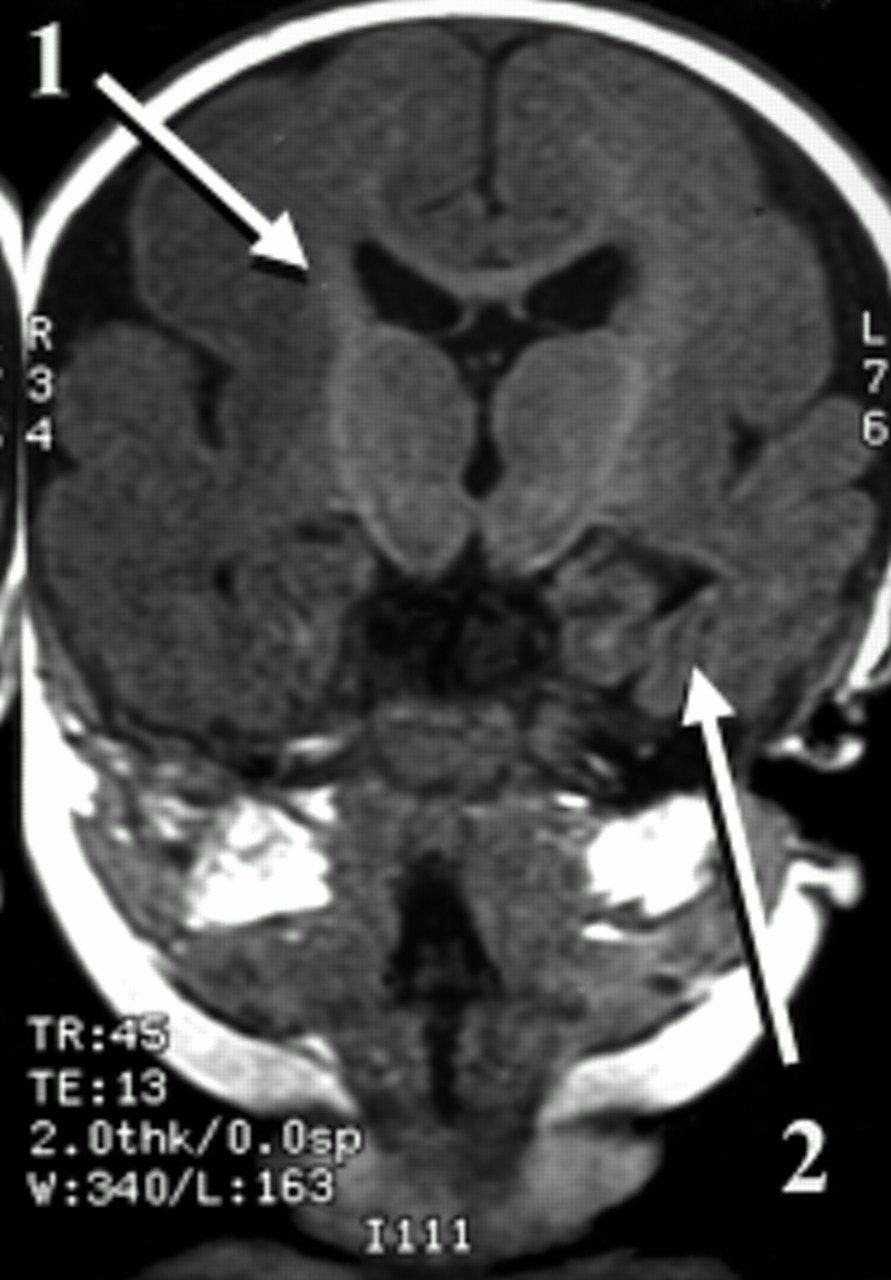
“True” callosal agenesis should be distinguished from secondary types which are associated with major malformations of the embryonic forebrain such as holoprosencephaly. Agenesis of the corpus callosum may be complete or partial. When there is complete absence there is no cingulate gyrus (fig 5). Associated enlargement of the occipital horns of the lateral ventricles is known as colpocephaly.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Agenesis of the corpus callosum. Sagittal T1 weighted magnetic resonance image. The arrow indicates the complete absence of the corpus callosum and the cingulate gyrus (compare to fig 1). In this 11 year old girl there was also dysgenesis of the right temporal lobe (not seen). She had learning difficulties and complex partial (temporal onset) seizures. Agenesis of the corpus callosum may be an isolated finding and there may be no associated developmental or neurological problems.
Isolated callosal agenesis may be inherited but no loci have been mapped and non-syndromic genetic transmission is rare. It has been associated with trisomy 18 and trisomy 13, and reported in more than 20 autosomal and many X linked malformation syndromes. Callosal agenesis is part of the fetal alcohol syndrome and is seen in lactic acidosis and non-ketotic hyperglycinaemia.
When agenesis of the corpus callosum is the only lesion there may be no symptoms although tests of perception and language may demonstrate disturbances of integration of hemispheric function. However, some patients have mental retardation, seizures or cerebral palsy.
Prenatal ultrasound allows diagnosis from 20 weeks gestation. When callosal agenesis is discovered on antenatal scan the prognosis is difficult to assess because the isolated lesion can be associated with normal development. A decision to terminate the pregnancy may depend on the demonstration of associated abnormalities.
Porencephaly
The term porencephaly is often used for any cavity in a cerebral hemisphere that commnunicates with a lateral hemisphere. However, it should probably be used only for circumscribed hemispheric necrosis that occurs in utero before the adult features of the hemisphere are fully developed. The relatively early development of these lesions is shown by their smooth walls and by associated developmental disturbances in the adjoining cortex such as polymicrogyria or distortion of the gyral pattern. This is relevant because unilateral or bilateral porencephalic cysts are found in children diagnosed as having cerebral palsy and there is often debate about the timing of the insult. Neuropathological texts debate whether or not there is a distinction between porencephaly and schizencephaly, and some cortical abnormalities do not fit neatly into any group (fig 4).
Schizencephaly
This term is used by radiologists to describe clefts which traverse the full thickness of the hemisphere, connecting the ventricle to the subarachnoid space. They are described as type I or “fused-lip” when the walls of the cleft are opposed, and type II or “open-lip” when cerebrospinal fluid separates the walls. Some of them are genetic—familial cases have been described and some sporadic cases are associated with mutations in the homeobox gene EMX2. The clefts are frequently bilateral and even when unilateral they are often combined with cortical dysplasia of the opposite hemisphere.
Epilepsy is common and sometimes the only problem is isolated partial seizures. There may be hemiplegia, quadriplegia, and learning difficulties of variable degree. If there is bilateral involvement of both opercular regions there may be facial apraxia and speech difficulties.
MALFORMATIONS OF POSTERIOR FOSSA STRUCTURES
At the end of the fourth week of gestation the neural tube divides into the three primary brain vesicles—the prosencephalon, the mesencephalon, and the rhombencephalon. The latter further subdivides into the metencephalon and the myelencephalon. The cerebellar hemispheres (neocerebellum) are derived primarily from the metencephalon, while the vermis (palaeocerebellum) is derived from the mesencephalon.
Malformations of posterior fossa structures include aplasia or hypoplasia of the cerebellar hemispheres (which may be combined with brainstem abnormalities). There may be abnormalities of the vermis, including the Dandy-Walker malformation (which consists of complete or partial agenesis of the vermis, dilatation of the fourth ventricle, and enlargement of the posterior fossa) and the Joubert syndrome (an autosomal recessive disorder characterised by absence or hypoplasia of the postero-inferior part of the vermis).
INVESTIGATIONS AND GENETIC COUNSELLING
Investigations
-
Magnetic resonance imaging is essential to delineate the anatomical abnormalities.
-
If a structural CNS abnormality is found consider chromosome analysis, especially if there are developmental problems or learning difficulties.
-
Further investigations will depend on the specific diagnosis. Metabolic disorders can cause structural abnormalities in the developing CNS.
-
Mutation analysis of specific genes may confirm a clinical diagnosis. Fluorescent in situ hybridisation (FISH) studies may detect microdeletion syndromes such as the Miller-Dieker syndrome.
-
Take blood to extract and store DNA or establish a lymphoblastoid cell line if no precise diagnosis can be reached, especially if life expectancy is short. Frozen tissue for DNA can be obtained when postmortem examinations are being performed.
Risk assessment
-
It is important to construct a three generation family tree, if necessary with examination and investigation of close relatives for subtle expression of a disorder, or evidence of carrier status.
-
The aim is to establish a diagnosis and give an accurate risk. If it is not possible to identify the precise aetiology it is usually possible to offer an empiric recurrence risk after examination of the parents.
Prenatal diagnosis
-
Prenatal diagnosis and termination of affected pregnancies is only one of a range of reproductive options open to parents, but for many couples it is the option of choice.
-
For the majority of developmental disorders of the nervous system, pre-implantation genetic diagnosis is not yet feasible.
-
For a condition following mendelian inheritance the option of donor gametes could be discussed.
-
For a condition with a strong environmental component it is imperative that measures are taken to minimise the risk of exposure in a future pregnancy. For neural tube defects, periconceptual supplementation with high dose folate has been shown to reduce the risk of recurrence in future pregnancies (see above).
-
When a specific diagnosis has been made and a chromosomal anomaly, genetic mutation or biochemical defect has been identified it is usually possible to offer prenatal diagnosis by chorion villus sampling at 11 weeks gestation in a future pregnancy. If this is not the case, detailed ultrasound scanning may be helpful in some instances.
-
Providing accurate genetic advice about developmental anomalies of the nervous system is a challenging task. Referral for specialist advice is strongly recommended.
Learning points
-
Neural tube defects (NTDs) are some of the most common congenital abnormalities of the CNS, although their prevalence in the UK has fallen. Nevertheless it is still important to counsel women of childbearing age about the need to take dietary supplements containing folate before becoming pregnant
-
There is an increased risk of NTDs in pregnant women who are taking certain drugs
-
Even small midline abnormalities over the spinal cord should be taken seriously in case there is underlying spinal dysraphism
Learning points
-
When assessing children the head circumference should be measured and plotted on a centile chart. The current measurement should be compared with previous measurements to determine whether or not there has been a progressive change with time
-
Some children with non-progressive abnormalities of head size are normal in their development, but children with micro- or macrocephaly and abnormal development should probably have magnetic resonance imaging of the brain
Learning points
-
The lissencephalies cause major developmental problems and may shorten life
-
In type II lissencephaly the cerebral malformations may be associated with eye or muscle abnormalities so that a comprehensive assessment is necessary
-
Some of the less severe migrational and organisational abnormalities may be clinically silent. However they may be associated with learning difficulties or seizures. Many will be discovered on magnetic resonance imaging of the brain. However some are microscopic, which is relevant in the assessment of some patients with refractory epilepsy
Learning points
-
Combined malformations may be found in infants with severe developmental and neurological problems
-
However, they may be present in patients with relatively minor learning difficulties or motor disability. Patients with agenesis of the corpus callosum may have no neurological problems. It is important to be guarded about the prognosis when such abnormalities are found on scanning in early life