Article Text

Abstract
Background Omphalocele is a congenital birth defect characterised by the presence of internal organs located outside of the ventral abdominal wall. The purpose of this study was to identify the underlying genetic mechanisms of a large autosomal dominant Caucasian family with omphalocele.
Methods and findings A genetic linkage study was conducted in a large family with an autosomal dominant transmission of an omphalocele using a genome-wide single nucleotide polymorphism (SNP) array. The analysis revealed significant evidence of linkage (non-parametric NPL = 6.93, p=0.0001; parametric logarithm of odds (LOD) = 2.70 under a fully penetrant dominant model) at chromosome band 1p31.3. Haplotype analysis narrowed the locus to a 2.74 Mb region between markers rs2886770 (63014807 bp) and rs1343981 (65757349 bp). Molecular characterisation of this interval using array comparative genomic hybridisation followed by quantitative microsphere hybridisation analysis revealed a 710 kb duplication located at 63.5–64.2 Mb. All affected individuals who had an omphalocele and shared the haplotype were positive for this duplicated region, while the duplication was absent from all normal individuals of this family. Multipoint linkage analysis using the duplication as a marker yielded a maximum LOD score of 3.2 at 1p31.3 under a dominant model. The 710 kb duplication at 1p31.3 band contains seven known genes including FOXD3, ALG6, ITGB3BP, KIAA1799, DLEU2L, PGM1, and the proximal portion of ROR1. Importantly, this duplication is absent from the database of genomic variants.
Conclusions The present study suggests that development of an omphalocele in this family is controlled by overexpression of one or more genes in the duplicated region. To the authors' knowledge, this is the first reported association of an inherited omphalocele condition with a chromosomal rearrangement.
- Omphalocele
- autosomal dominant
- 1p31.3 duplication
- genome-wide linkage
- academic medicine
- cell biology
- complex traits
- copy-number
- genome-wide
- reproductive medicine
- molecular genetics
- clinical genetics
- genetic epidemiology
- linkage
- genetics
Statistics from Altmetric.com
- Omphalocele
- autosomal dominant
- 1p31.3 duplication
- genome-wide linkage
- academic medicine
- cell biology
- complex traits
- copy-number
- genome-wide
- reproductive medicine
- molecular genetics
- clinical genetics
- genetic epidemiology
- linkage
- genetics
Introduction
Omphalocele (MIM 164750) includes a group of birth defects known as ventral wall defects. Omphalocele affects ∼1 in 5000 live births1 with a higher ratio of males to females, and is due to abnormal closure of the abdominal wall. During the sixth to 10th week of fetal growth the intestines grow and project from the abdomen into the umbilical cord. If the intestine fails to return to the abdomen during the 11th week of pregnancy, improper closure of the abdominal wall occurs generating an omphalocele. An omphalocele is covered by a clear sac or membrane through which the umbilical cord is inserted. The size of the omphalocele varies from small (including the small intestine) to very large, containing most of the abdominal organs, such as the liver and small and large intestine. A ‘giant omphalocele’ is seen in around 1 in 10 000 births and occurs when most of the liver protrudes into the defect. An omphalocele can occur in association with over two dozen syndromes2–4 including Beckwith–Wiedemann syndrome (MIM 130650), otopalatodigital syndrome type II (MIM 304120), Melnick–Needles syndrome (MIM 309350), lethal omphalocele-cleft palate syndrome (MIM 258320), and Shprintzen omphalocele syndrome (MIM 182210).
Although the aetiology of an omphalocele is unknown,5 isolated omphaloceles are suspected to be multifactorial, with considerable evidence for genetics playing a role. Various families have been reported with multiple affected members with omphalocele consistent with autosomal dominant,6–8 autosomal recessive, and X-linked inheritance9 10 with or without other associated anomalies. Infants with an omphalocele have an increased incidence of other abnormalities involving the gastrointestinal tract11 (stomach and intestines), cardiovascular system (heart),12 genitourinary tract (kidneys and bladder), musculoskeletal system, central nervous system (brain and spinal cord), and the limbs. Many infants born with an omphalocele are premature. There is also a reported association between younger paternal age and an omphalocele.13 14 Chromosomal anomalies are also reported in fetuses with an omphalocele, including trisomy 13, 18, and 21.3 15 16 Although no single gene mutations are known to cause non-syndromic omphalocele, syndromes such as Donnai–Barrow syndrome and Townes–Brocks syndrome associated with omphalocele and an umbilical hernia are caused by mutations in the LRP2 gene on chromosome 2q23.3-31.1,17 and the SALL1 gene on chromosome 16q12.1,18 respectively.
Here, we report a genome-wide linkage analysis in a large Caucasian family (UR0114) with non-syndromic omphalocele and an autosomal dominant mode of inheritance.6 This analysis provides significant evidence for a susceptibility locus in a genomic region of 2.74 Mb on chromosome 1p31.3. Molecular characterisation of the linked genomic region revealed a 710 kb genomic duplication including seven known genes in all affected individuals.
Methods
Description of the family (UR0114)
A five-generation non-syndromic omphalocele family with autosomal dominant inheritance and incomplete penetrance was first reported by Kanagawa et al.6 The family was recently revisited and extended with five additional individuals. The family comprises 28 individuals, including nine (seven males and two females) members with the omphalocele phenotype. All affected members required surgical intervention during the first days of life to correct the omphalocele and to close the abdominal wall defect. There were no other birth defects identified or associated with the omphalocele in the affected family members. Informed consent was obtained from all subjects who participated in the study, and blood samples were collected from all cooperating family members. Samples from a total of 17 individuals were used for the linkage analysis: eight affected individuals, and nine unaffected individuals (figure 1).

Pedigree of family UR0114 with an omphalocele. Affected individuals are shown with black symbols and normal individuals are shown with clear symbols. Individuals with a slash across their symbol are deceased. The status is unknown for individuals 11 and 12, with grey. Individual 22 in the family was apparently affected. Individuals numbered under each symbols were used in the linkage analysis. Genotypes and haplotypes of chromosome 1p31.3 single nucleotide polymorphism markers are shown below selected individuals of family UR0114. Haplotypes associated with affected status are shown in red. Haplotype analysis indicated that the co-segregating segment of the omphalocele locus is flanked proximally by marker rs2886770 and distally by marker rs1343981 on chromosome 1p31.3.
Genome-wide scan and haplotype analysis
For the genome-wide linkage scan, we used Affymetrix (Santa Clara, California, USA) GeneChip Mapping EA 10K XbaI arrays containing 10 555 single nucleotide polymorphisms (SNPs). These SNP markers are evenly distributed across the genome, with a mean intermarker distance of 250 kb and an average heterozygosity of 0.38 (Affymetrix). The assay was performed using 250 ng of genomic DNA, and 99% of the SNPs were determined unequivocally for each sample. Scanned images were processed with the Affymetrix Micro Array Suite, and data analysed with GDAS v2 software. PedCheck was used for the detection of Mendelian incompatibility of genotypes.19 Twenty-five informative SNP markers were used for haplotype reconstruction and analysis.
SNP genotyped data were imported into the linkage-analysis programs GENEHUNTER20 and Mendelian errors were identified by MERLIN.21 In the initial genome scan, evidence of linkage was assessed with a non-parametric, penetrance-independent, affected-only, and allele-sharing analysis (NPL score). With use of MERLIN, one can convert this into a non-parametric logarithm of odds score (LOD) by maximising the likelihood with respect to a scalar parameter, d, that measures the amount of excess sharing of identical-by-descent alleles among affected relatives corresponding to the null hypothesis dp0 of no linkage. We used the SallL scoring function20 22 and the exponential allele sharing model to generate the relevant linkage statistic.
Whole genome array comparative genomic hybridisation
Isothermal oligo design, array fabrication, DNA labelling, array comparative genomic hybridisation (aCGH) experiments, data normalisation and log2 (Cy3/Cy5) ratio calculations were performed by NimbleGen (Madison, Wisconsin, USA). The arrays were constructed by maskless array synthesis technology (NimbleGen Systems, Inc.), with 385 000 oligonucleotides being synthesised by photolithography on an array by previously described methods.23 24
Quantitative microsphere hybridisation
Genomic copy number determination was done with quantitative microsphere hybridisation (QMH) essentially as described previously.25 A series of seven unique sequence (patent pending) test probes specific to chromosome 1p31.3 were designed, as well as a disomic reference probe for β actin (ACTB) on chromosome 7p22. Probes were selected based on their unique sequence composition, lack of secondary structure, and optimum Tm. Each probe was synthesised using a 5′-C, 6-amino modification (Integrated DNA Technologies, Coralville, Iowa, USA) for coupling to spectrally distinct carboxylated microspheres via a carbodiimide coupling reaction as described previously.25 26 Quality control procedures for each microsphere conjugated probe were performed to ensure proper attachment of probes to microspheres. Genomic templates were prepared with 50 ng of fragmented biotin labelled genomic DNA sample per each QMH reaction. Hybridisation reactions included chromosome 1 probes HOOK1, CHR1-63, FOXD3, ITGB3BP, ROR1, CHR1-64.5, CHR1-65, as well as the disomic reference probe ACTB. After analysis of each sample by flow cytometry, the ratios of geometric mean fluorescence intensities (MFI) of test probe to reference probe were calculated and statistics (eg, mean, 95% CIs, and SD) were computed. Geometric MFI values were previously shown to be the most accurate for assessing data from QMH assays since data are collected in logarithmic mode on the flow cytometer.25 27
Results
Linkage
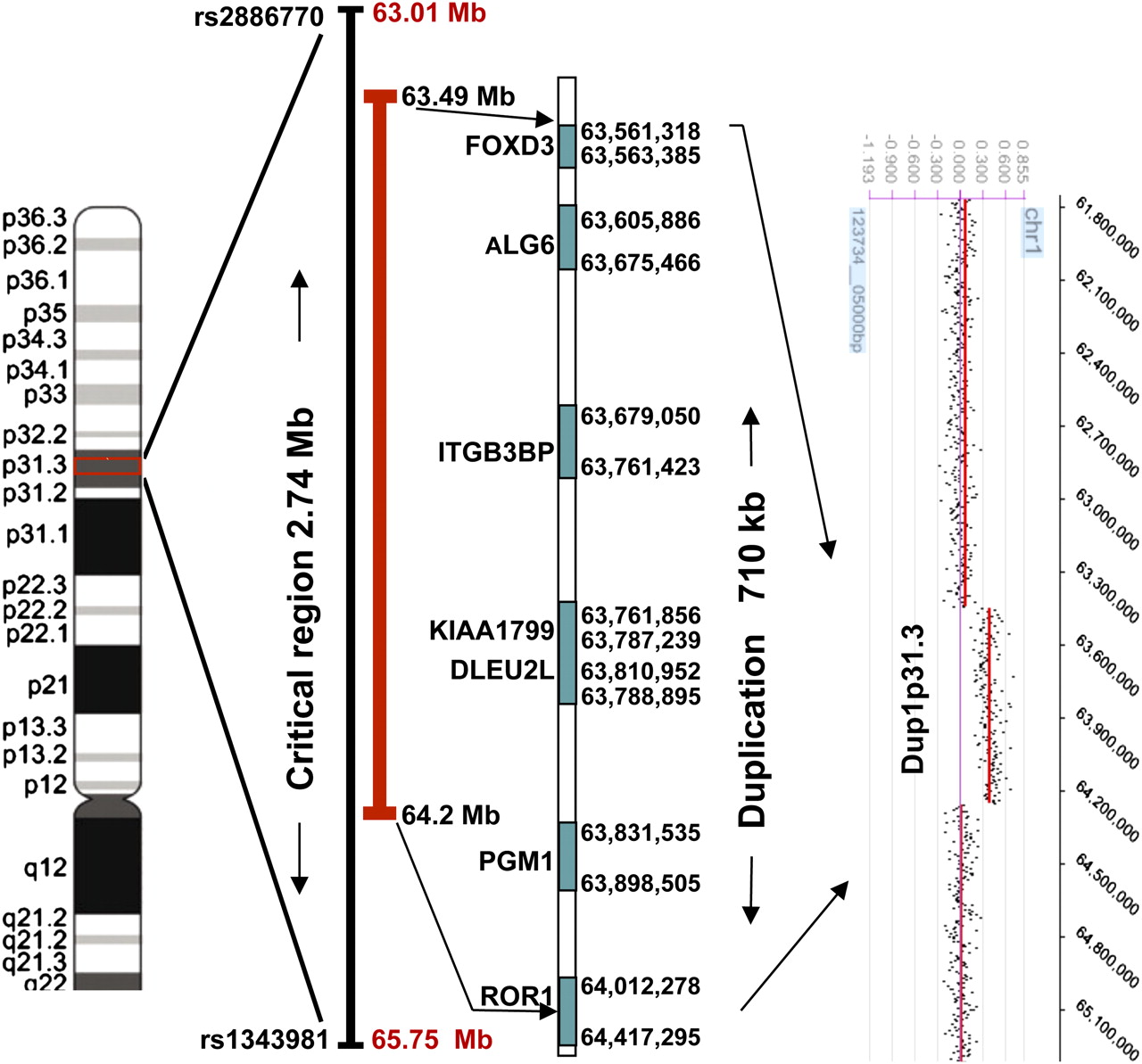
The genome-wide linkage scan with the use of an NPL analysis provided significant evidence of linkage for an omphalocele locus at marker rs937805 on chromosome 1p31.3 (NPL=6.92 and p=0.0001; non-parametric LOD=2.70 and p=0.0001). These data were also supported by subsequent parametric linkage analysis, with a dominant mode of inheritance and reduced penetrance (99%). Eleven SNP markers, spanning a region of 2.74 Mb (rs2886770–rs1343981), showed non-parametric LOD scores of 6.9 (table 1). No additional peaks with statistically significant LOD scores were found in the genome (figure 2). Haplotype analysis for the 1p31.3 linked region was performed using 13 informative SNP markers. Informative crossovers in multiple affected individuals 6011, 6014, and 6015 between SNP markers rs2886770/rs1411391, and a single informative crossover in affected individual 6016 between rs1327118/rs1343981, defined the omphalocele candidate region of 2.74 Mb, bordered by proximal marker rs2886770 (chr1: 63014807) and distal marker rs1343981 (chr1: 65757349) (figure 3).
Single nucleotide polymorphism (SNP) markers data in the candidate linked loci on 1p31.1 in family UR0114

Multipoint linkage analysis using NPL in the genome-wide scan on family UR0114 with an omphalocele. The x-axis (shown on top) represents the chromosomal orientation of 22 autosomes and sex chromosomes, and the y-axis represents the parametric LOD score.

Schematic representation of an omphalocele duplicated region on chromosome 1p31.3. The omphalocele critical region is 710 kb in 1p31.3. This region contains FOXD3, ALG6, ITGB3BP, KIAA1799, DLEU2L, PGM1, and proximal portion of ROR1 genes and duplicated boundary markers. Array comparative genomic hybridisation defines the duplication interval on 1p31.3.
Whole genome aCGH and QMH
Oligonucleotide aCGH showed a duplication of ∼710 kb genomic region at 1p31.3. This observation was confirmed by QMH analysis. The QMH assay was used to test all omphalocele subjects used in the linkage analysis. All seven QMH probes plus a control probe were included in each multiplex QMH reaction. The aCGH indicated that the proximal boundary of the duplicated region is located at 63.5 Mb, proximal to FOXD3. The distal border of the duplication is located within the ROR1 gene (64.2 Mb). This duplicated 710 kb segment was present in all affected individuals (6002, 6004, 6006, 6009, 6011, 6014, 6015, and 6016) but was absent in two normal individuals (6001 and 6017) who shared the affected allele (figure 4). Additionally, to test this finding, we have genotyped DNA samples of all individuals using three polymorphic microsatellite markers (D1S2835, D1S348, and D1S515) on chromosome 1p31.3 region. Marker (D1S348) within the duplicated region produced three alleles in all those affected and two alleles for normal. Markers D1S2835 and D1S515, that are outside the duplication but within the linked region, gave two alleles for all analysed individuals (supplementary figure 5). Multipoint linkage analysis using the duplication as a marker gave a maximum LOD score of 3.2 at 1p31.3 under the dominant model. The best-fitted parametric model that was used for obtaining the optimum linkage results was a dominant model with 99% penetrance and a disease-allele frequency of 0.0001.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Quantitative microsphere hybridisation analysis showing relative copy number changes at 1p31.1. Graphs showing results of quantitative assays for selected individuals listed in the text. The points indicate the normalised and duplicated DNA copy number for various genes at this location for a particular assay. The grey line at 1.0 indicates normal copy number.
Discussion
Although there have been a number of studies undertaken to identify the genetic variations responsible for omphalocele, no single susceptibility gene has been identified to date that plays a major role in this disorder.28 Here, we report the results of the first genome-wide linkage scan and other genetic analysis, providing significant evidence of a novel omphalocele susceptibility locus on chromosome 1p31.3 in a large pedigree (UR0114). The study further demonstrates duplication of six contiguous genes on chromosome 1p31.3 as the cause of omphalocele in this family, signifying that extended families are vital for efficient linkage based disease gene identification. Haplotype analysis with critical recombination events allowed us to initially define a 2.74 Mb disease interval. Haplotype analysis identified the risk haplotypes shared by all affected individuals that was not found in any of the unrelated unaffected spouses, but was shared by two normal individuals (6001 and 6017). The disease locus was shown to be non-recombinant in all affected individuals and was found to have originated from the affected great-grandfather (individual 22) who was not available for analysis.
We have discovered that individual 22 may have been affected, although he was reported previously as unaffected.6 While re-interviewing family members, we discovered that individual 22 had undergone previous abdominal surgery and had scars, though no diagnosis of an omphalocele was known to his children. Multiple family members can recall the appearance of the scars throughout his life, which are typical of an abdominal wall defect repair. In light of the genetic findings, the patient's history is most consistent with a congenital abdominal wall defect and individual 22 fathered two affected sons (6002 and 6009) through two different unaffected, unrelated spouses. The affected sons later transmitted the mutant allele to six others who were affected in two generations. The 710 kb duplicated genomic region which is shared by all those affected is non-recombinant and is the same in all affected individuals, but was not found in two normal individuals 6001 and 6017 who shared the affected allele. These data suggest that a gene dosage effect is the most likely pathogenetic mechanism for omphalocele phenotype in this family. The omphalocele in this family demonstrates high penetrance, in that all unaffected parents and normal subjects were negative for the duplication. The haplotype data also support the autosomal dominant mode of inheritance in this family. This is well corroborated by our subsequent linkage analysis using the duplication as a genetic marker in which the LOD score increased from 2.7 to 3.2 under the same dominant model. The duplication is absent in the database of genomic variants (http://projects.tcag.ca/variation/). This catalogue currently comprises data collected from 42 different studies of copy number variation (CNV) assayed in >3000 individuals. Given the large size of the CNV we identified, it is well within the detection limits of most techniques used for the detection of CNVs; therefore, the fact that this 1p31.1 duplication has not been detected in >3000 controls demonstrates that the duplication we report is not a common variant segregating in the population. In addition, we have tested 250 sporadic omphalocele samples by quantitative PCR (qPCR), but did not find any evidence for a CNV of the chromosome 1 linkage region in any of them. However, none of these individuals was tested for point mutations in any of the candidate gene coding regions and/or splice junction variants.
The 710 kb duplicated genomic region on chromosome 1p31.3 band contains seven known transcripts, which therefore represent candidates for omphalocele. These include forkhead box D3 (FOXD3, MIM 611539), asparagine-linked glycosylation 6, α-1, 3-glucosyltransferase homologue (ALG6, MIM 604566), integrin β-3 binding protein (ITGB3BP, MIM 605494), phosphoglucomutase 1 (PGM1, MIM 171900), receptor tyrosine kinase-like orphan receptor 1 (ROR1, MIM 602336), deleted in lymphocytic leukaemia 2-like (DLEU2L), and KIAA1799 (figure 3). We hypothesise that over-expression of one or a combination of these genes is likely to be responsible for omphalocele in these affected individuals.
FOXD3 is a member of the forkhead box transcription factor family, and these genes play a fundamental role in development and are responsible for embryo patterning and neural development. FOXD3 is especially shown to play an important role in mesoderm29 and neural crest development.30 Moreover, FOXD3 is known to interact with other developmental genes including the NODAL,31 BMP1, and WNT signalling32 pathways. Thus, over expression of FOXD3 could conceivably contribute to the mesodermal abnormalities observed in omphalocele. Interestingly, ALK3 is one of two BMP1 receptors that act in the BMP signalling pathway, and the Alk3-knockout mouse exhibits severe defects of secondary ventral body wall formation, replicating the omphalocele phenotype in human.33
Similarly, ALG6, a homologue of yeast glycoprotein glycosyl transferase (ALG6) is known to be mutated in human congenital disorders.34–36 Similar to FOXD3, ALG6 overexpression may disrupt the fine tuning of embryo development. ITGB3BP is a transcriptional regulator of the integrin family that interacts with NRIF337 and cyclin A38 and induces rapid and profound apoptosis in breast cancer cells.39 PGM1 or phosphoglutamase1 polymorphism is associated with diabetic pregnancy40 and body mass index.41 The ROR1, orphan receptor tyrosine kinase 1, is known to play a critical role in heart and bone development42 and has been shown to play a vital role in mouse development.43 Moreover, it is involved in the WNT signalling pathway.44 In addition, ROR2 gene mutations are responsible for Robinow syndrome, a congenital short stature syndrome with facial and genital defects whereby umbilical anomalies are reported in 20% of affected subjects.45 46 The other two genes in the duplicated region, KIAA1799 and DLEU2L, are less well characterised.
The six known duplicated genes with their presumed overexpression may be involved in the omphalocele phenotype in the present family and partial duplication of ROR1 represents an additional genetic mechanism for the phenotype. This evidence supports the proposal that the duplicated region disrupts the fine tuning of gene expression in embryo development, and the lack of this regulation gives rise to the spectrum of abnormal developmental phenotypes and needs further exploration. Identification of other individuals with omphalocele who present with overlapping genomic rearrangements may refine a critical region in 1p31.3, allowing more precise definition of a dosage sensitive locus underlying this phenotype.
Chromosome 1p interstitial duplications are rare, and no cytogenetically visible chromosomal rearrangements of 1p31.3 have been reported in association with omphalocele. An interstitial dup (1p) involving the p32-p21.2 region associated with severe intrauterine growth retardation and an inguinal hernia was reported by Dhellemmes et al.47 Direct duplication of chromosome 1, dir dup(1) p21.2-p32 with multiple congenital anomalies including an inguinal hernia was also reported.48 Similarly, a girl has been described with multiple congenital anomalies, including an omphalocele but with a diploid/tetraploid karyotype and mosaicism for a translocation involving chromosome band 1p32 [46,XX,t(1;6)(p32;q13)].49 Halal et al50 also described translocations between chromosomes 1 and 2, along with a duplication of chromosome 1p in which several congenital anomalies including an umbilical hernia were reported.
The present exceptional large multigenerational family with an autosomal dominant form of omphalocele provides evidence of linkage to 1p31.3, and also provides evidence specifically for overexpression of one or more genes within the duplicated 710 kb region causing the phenotype in this family. This is the first report to elucidate the pathogenic mechanism and molecular location of gene(s) involved with development of omphaloceles.
Web resources
The URLs for data presented herein are:
Affymetrix, http://www.affymetrix.com/products/arrays/specific/10k.affx
Ensemble, http://www.ensembl.org/
Genome Database, http://www.gdb.org/
NCBI (Build 35.1), http://www.ncbi.nih.gov/
The disease identifiers for the OMIM http://www.ncbi.nlm.nih.gov/Omim/ (for FOXD3, ALG6, ITGB3BP, KIAA1799, DLEU2, PGM1, ROR1).
The Ensembl Gene ID (http://www.ensembl.org/) discussed in this paper are: FOXD3 (ENSG00000187140), ALG6 (ENSG00000088035), ITGB3BP (ENSG00000142856), ROR1 (ENSG00000185483), PGM1 (ENSG00000079739).
Acknowledgments
We are grateful to the patients and their families for their cooperation in the study.
References
Footnotes
UR and SKN equally contributed.
Funding The study was supported in part by Green Cross Blood Bank, Ahmedabad, Gujarat, India. SKN is supported by Oklahoma Medical Research Foundation institutional grant 9124, for linkage analysis. KM is supported by a GIS-Institut des Maladies Rare grant. AJS is supported by funding from the European Community's Seventh Framework Program under grant agreement 219250.
Competing interests None.
Patient consent Obtained.
Ethics approval Ethics committee of University of Nebraska Medical Center, Nebraska, USA.
Provenance and peer review Not commissioned; externally peer reviewed.