Article Text

Statistics from Altmetric.com
Congenital duodenal obstruction (CDO) is a relatively common neonatal anomaly with a reported incidence of 1 in 2500–10 000 live births.1 2 Diagnosis can be made prenatally with ultrasound (US) during antenatal care or during the postnatal period commonly as bilious vomiting and feeding intolerance. Patients with a partial stenosis can survive to present in a much-delayed fashion.3–6
Annular pancreas (AP) is a rare congenital disorder that may be clinically asymptomatic, may present as a neonatal intestinal obstruction or as a more complex pathology in adults. The incidence is reported as 1–3 in 20 000. AP may be asymptomatic7 and discovered incidentally during routine investigations for other conditions or at autopsy. Most of the cases are diagnosed either prenatally or in the first few days of life.8 If the condition is neither diagnosed prenatally nor presents with complications in early life, it may be undetected until adulthood with varying complications, such as pancreatitis or gastric outlet obstruction, thereby making the diagnosis difficult. AP may coexist with other congenital anomalies.3 8–11 We report a case of obstructive jaundice from choledocholithiasis due to an AP that caused duodenal stenosis and biliary stasis.
A 12-year-old girl who was referred from western Uganda presented with postprandial abdominal pain, food fear, progressive weight loss, jaundice and dark urine for 2 years. She reported often missing school due to abdominal pain. Initial US obtained by her local care providers showed dilated intrahepatic and extrahepatic ducts, a distended gall bladder and a stone in the common bile duct (CBD). The patient weighed only 32 kg and appeared wasted with moderate jaundice and scleral icterus during her physical exam. Her abdominal examination was unremarkable. Her initial laboratory workup showed mild anemia, no leukocytosis, significant hyperbilirubinemia with mildly elevated liver enzymes and a negative infectious workup (table 1).
Pre-operative laboratory work-up
She had a cardiac echocardiogram that was normal and a repeat abdominal US that again demonstrated dilated CBD to 2 cm, dilated intrahepatic and extrahepatic ducts, a distended gall bladder and an obstructing stone in the CBD measuring 2.3×1.5 cm. Basing on these findings, the team elected to proceed with an exploratory laparotomy. Owing to resource limitations, magnetic resonance cholangiopancreatography (MRCP), endoscopic retrograde cholangiopancreatography (ERCP) and intraoperative fluoroscopy were not available options.
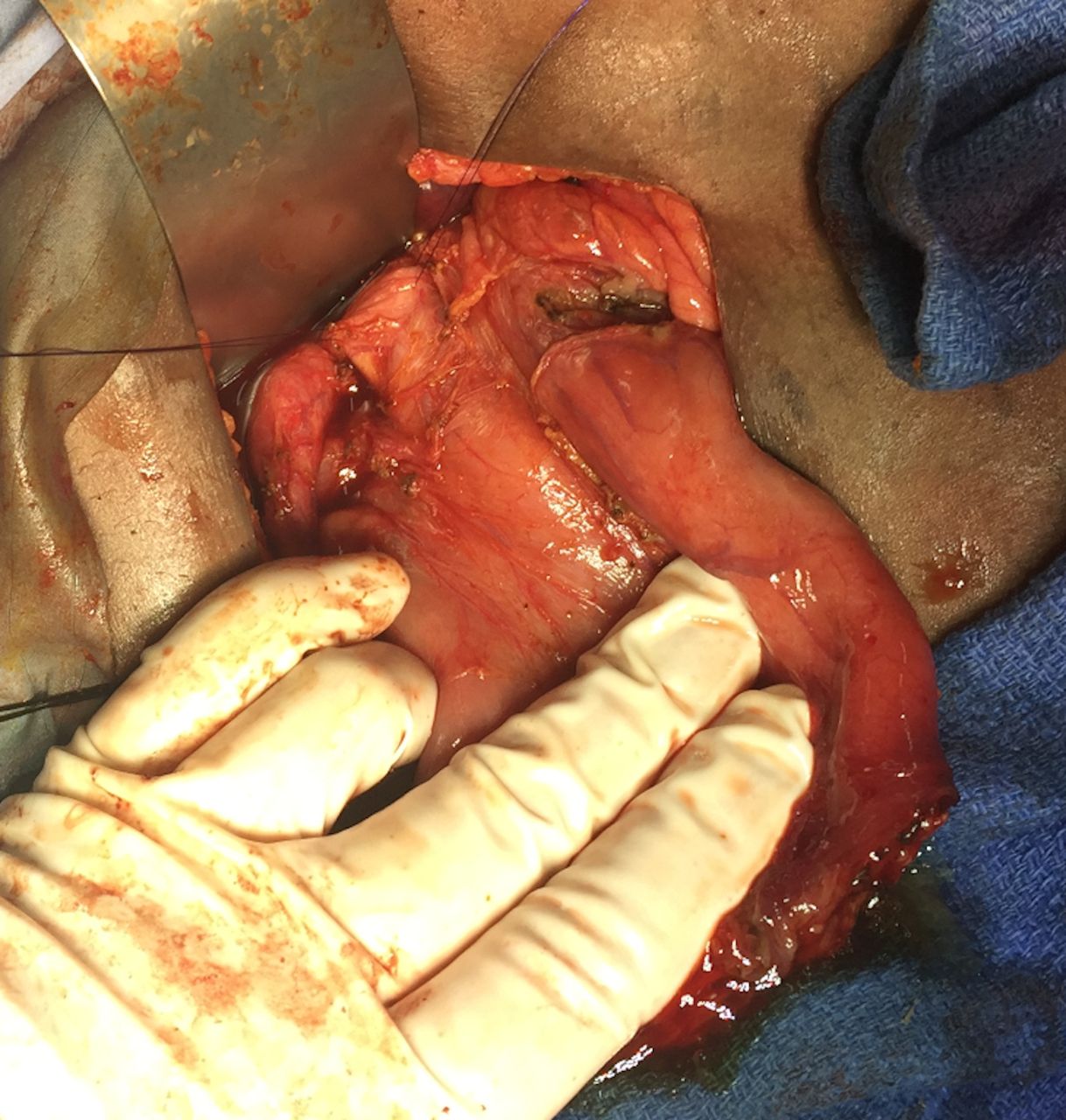
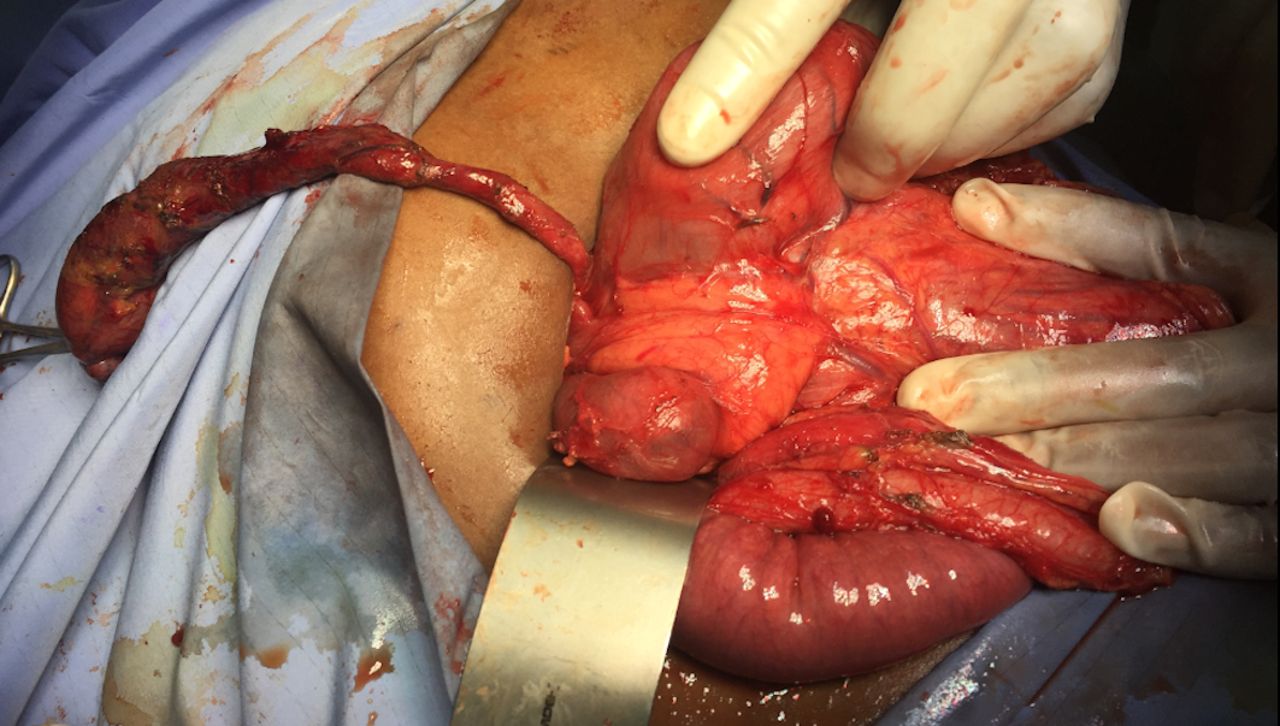
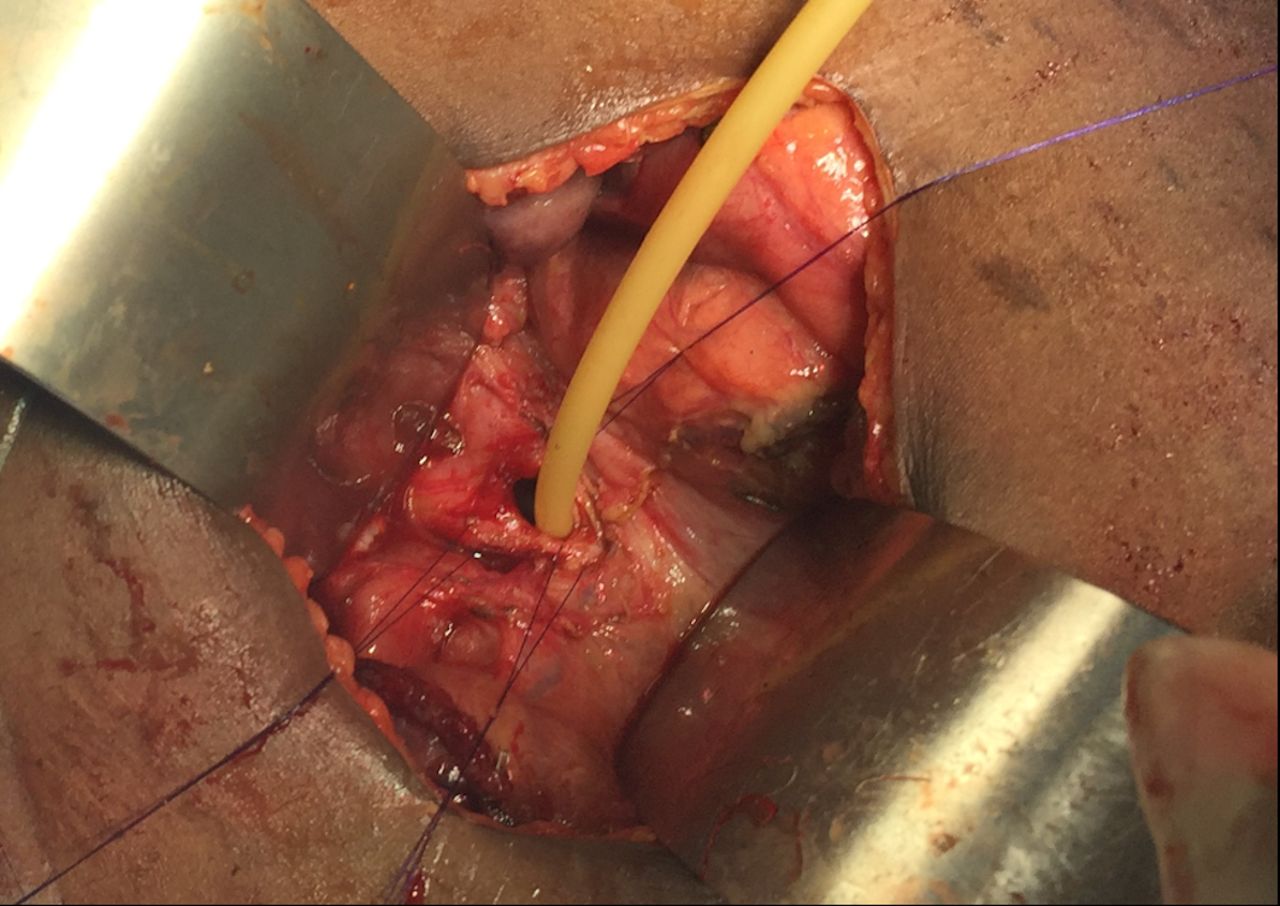
Surgery commenced through a right subcostal incision and intraoperative findings demonstrated a dilated gall bladder, cystic duct and bile duct (2 cm) with a palpable stone. The proximal duodenum also appeared very dilated with a patulous pylorus and a decompressed distal duodenum. After medializing the ascending and transverse colon, the duodenal obstruction was identified with an associated AP. The duodenum was adequately mobilized and freed from its retroperitoneal attachments via a Kocher maneuver. Attempted flushing of the stone through the cystic duct was unsuccessful. The CBD was therefore opened, and the stone was extracted. The residual debris was flushed, and a T-tube was placed. A diamond shaped duodenoduodenostomy was then performed with tapering of the proximal duodenum to prevent anastomotic dysfunction. The patient’s recovery was uneventful at short-term follow-up (figures 1–5).
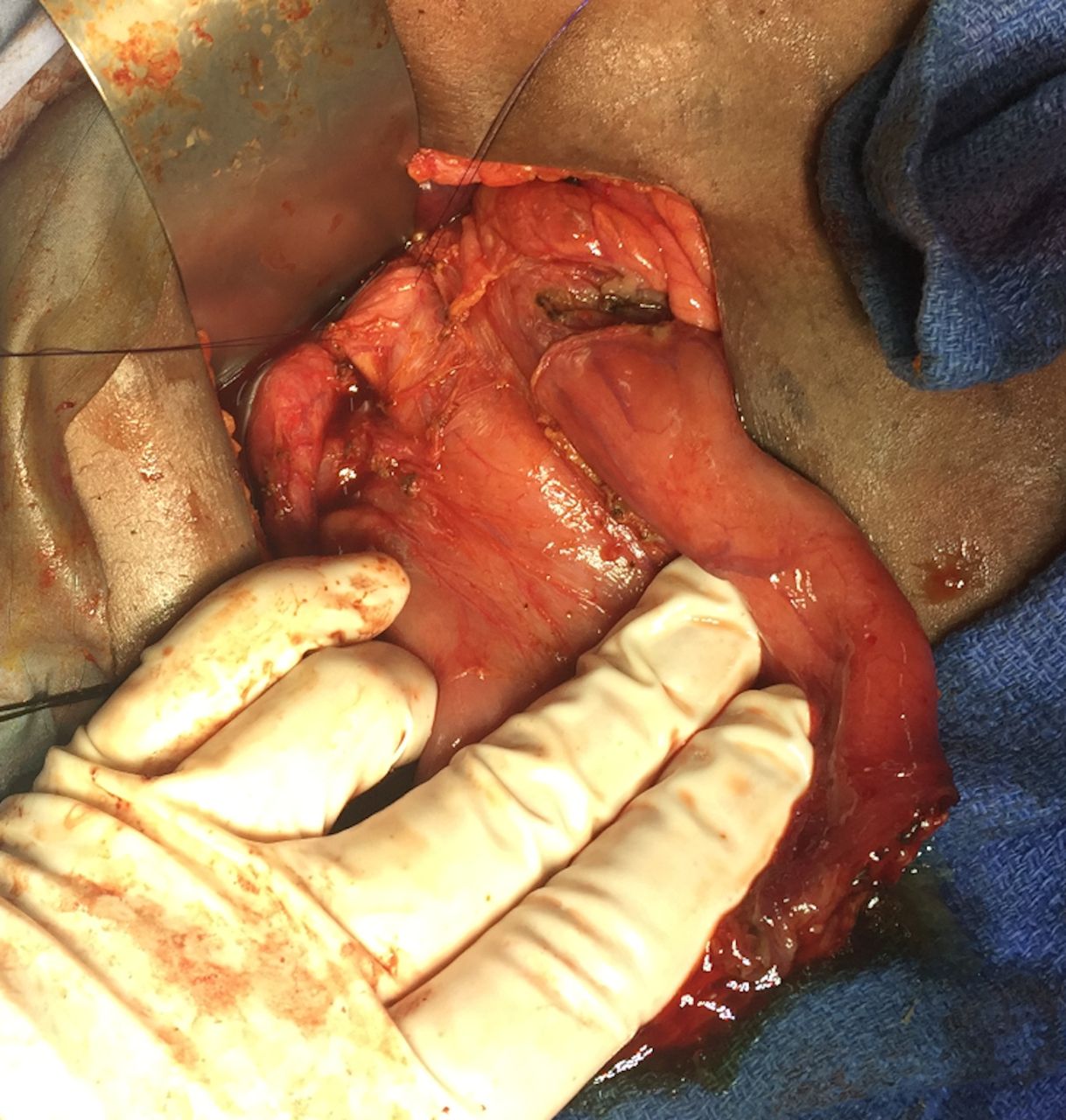
Bile duct and palpable stone.

Dilated duodenum, patulous pylorus, decompressed distal duodenum, annular pancreas with duodenal obstruction.
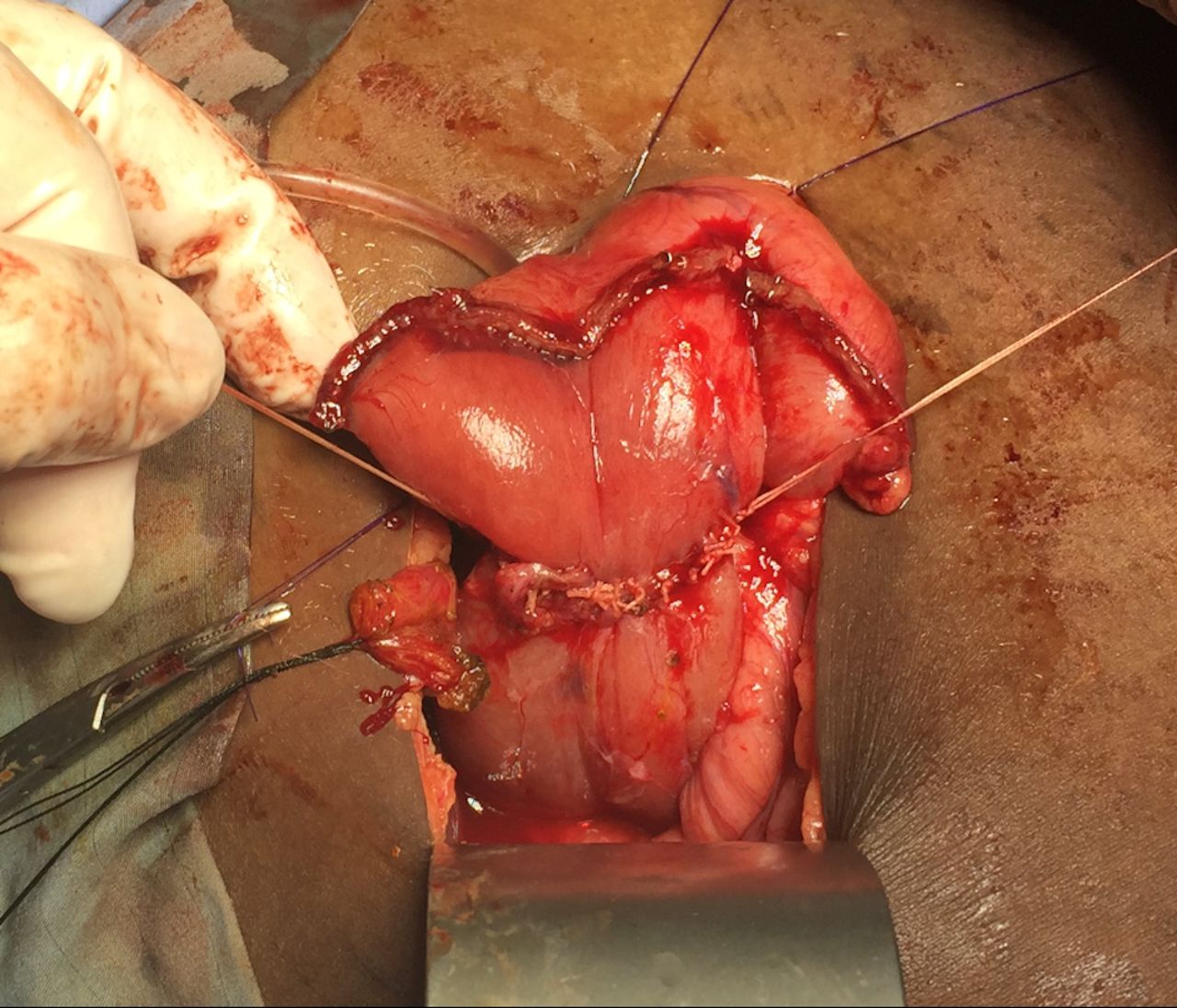
Duodenoduodenostomy with tapering of proximal duodenum.
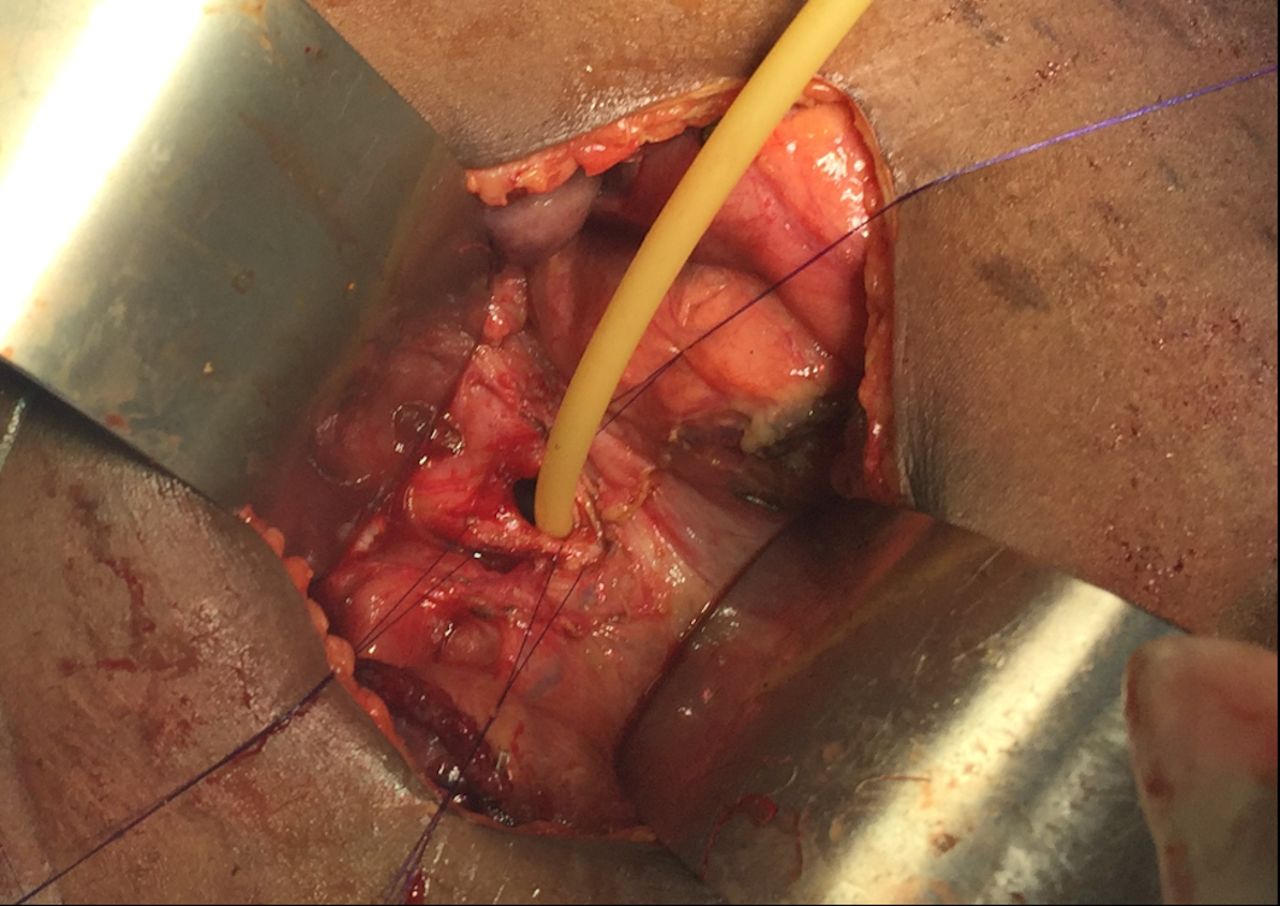
T-tube placement.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
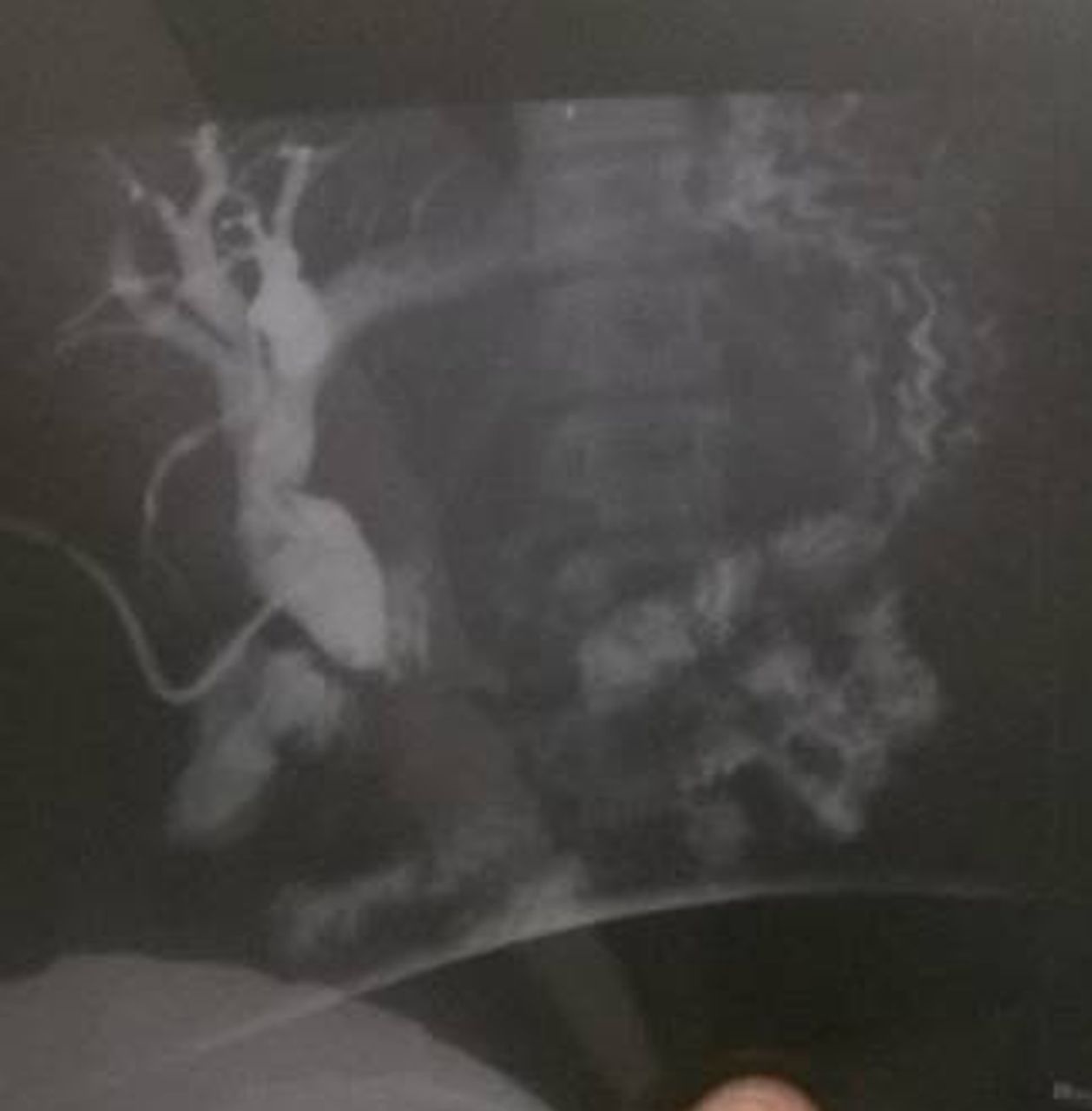
Cholangiograms after clamping T-tube for 8 weeks, 3.5 months postoperative, asymptomatic, no jaundice and normal bilirubin levels. Drain was thereafter removed.
The causes of CDO may be intrinsic or extrinsic. Intrinsic causes may include duodenal atresia, stenosis or web. Extrinsic obstruction may result from malrotation with Ladd’s bands, preduodenal portal vein or AP. Obstructions usually occur in the second part of the duodenum and are often associated with prematurity (46%) and maternal polyhydramnios (33%). There may be associated anomalies, including Down’s syndrome (>30%), malrotation (>20%), congenital heart diseases (20%) and other gastrointestinal (GI) tract and renal anomalies.3–6
AP is a congenital anomaly that can present in the neonatal period or can remain asymptomatic.12 Autopsy reports reveal an incidence of 5–15/100 000.13 ERCP has improved the diagnosis of AP with reports as high as 1 in 150 patients undergoing this procedure.14 AP was first described by Tiedemann in 1818 and was named by Ecker in 1862.15 These cases were generally found in patients who underwent surgery for duodenal atresia as an incidental finding as was the case in our patient.
Several theories have been documented to explain the embryological development of AP.8 13 16 17
AP typically presents in the neonatal period with intestinal obstruction, poor feeding, bilious vomiting (if proximal to the ampulla of Vater), non-bilious vomiting and irritability. There may be a history of polyhydramnios 16 18 if prenatal USs have been performed.
When children present in infancy or in early childhood, they have GI symptoms including poor feeding, vomiting and abdominal distention. Most of our patients in Uganda present late with malnutrition, failure to thrive, bile stained vomiting and—less frequently—abdominal cramps. Our patient presented with postprandial abdominal pain and fear of food. Adults are much more likely to describe upper abdominal pain or to present with features of pancreatitis.3 9 12 14
AP can be identified preoperatively using radiological and endoscopic studies including upper GI studies, CT, MRI, MRCP or ERCP. In more than 40% of cases, the diagnosis is made only during laparotomy as was the case in our patient.13 19–21
US is the favored initial study for patients with abdominal symptoms and jaundice; however, ERCP is the procedure of choice in patients with biliary obstruction, especially if the US findings are equivocal.22 Our patient only had a US and did not have an ERCP prior to surgery owing to resource limitations. Endoscopic ultrasonography can also be useful in non-obstructive forms.13 23 24
There are few reports of AP presenting as obstructive jaundice in the literature, as most reports of biliary stasis are attributed to malignancies.14 This makes the present case one of the few encountered and the only case in this pediatric age group presenting with biliary obstruction in such a resource-limited environment.
The symptomatic, obstructing AP has been treated surgically. The preferred treatment is a bypass operation, such as gastrojejunostomy or duodenojejunostomy, but some authors have treated by division of the annulus with transverse duodenoplasty, duodenoduodenostomy or Whipple’s procedure, depending on the intraoperative findings.13 20 25
Previous attempts at resection or division of the annulus26 resulted in many complications, such as duodenal leak, pancreatitis and pancreatic fistula. The current practice is to bypass the annular constriction by duodenoduodenostomy or duodenojejunostomy from the proximal duodenal bulb to the jejunum.9 13 We performed a diamond-shaped duodenoduodenostomy with tapering of proximal duodenum to minimize anastomotic dysfunction and prolonged obstruction from dilated proximal duodenum,14 although proximal duodenal dilation usually resolves after relief of the obstruction.2
Obstructive jaundice may not only be caused by an AP (on long-standing obstruction from biliary stones can be found concurrently with AP). Other causes may include carcinoma of the ampulla of Vater, pancreaticobiliary maljunction, choledochal cyst, anomalous junction of the pancreatic and CBDs, aberrant biliary ducts, pancreas divisum, multiple communicating intrahepatic and extrahepatic duct cysts, Caroli disease, pancreaticobiliary abnormalities associated with situs anomalies and aberrant pancreatic ducts associated with enteric duplication cysts.14 27–29
The treatments are controversial and vary with the associated anomaly. They include Whipple’s procedure, pancreaticoduodenectomy, duodenoduodenostomy, portal jejunostomy transduodenal sphincteroplasty, duodenojejunostomy, gastrojejunostomy, subtotal gastrectomy and others.19 20 28
Close, long-term follow-up after treatment for AP is necessary because some patients develop complications, such as jaundice, upper GI motility disorder, failure to thrive and chronic diarrhea.30 We shall continue to follow up the patient and to request postoperative biochemistry on subsequent, long-term follow-up reviews.
This is a rare case of AP presenting as obstructive jaundice in an adolescent. The resource-limited setting presented unique challenges owing to limitations in available workup and treatment options.
Data availability statement
All data relevant to the study are included in the article or uploaded as supplementary information.
Ethics statements
Ethics approval
Ethical approval was obtained from Mbarara University research ethics committee (# 31/05–20).
References
Footnotes
Contributors FO, MC and DO contributed to conception and design, the acquisition of data or analysis and interpretation of data, drafting the article or revising it critically for important intellectual content, final approval of the version published, agreement to be accountable for the article and to ensure that all questions regarding the accuracy or integrity of the article are investigated and resolved. AO, AMN, NK, JS, and GV contributed to the acquisition of data or analysis and interpretation of data.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.