Article Text

Abstract
This report describes an 11 month old female baby with features of pentasomy X. A molecular and cytogenetic evaluation revealed that her karyotype was 49,XXXXX and her extra X chromosomes were of maternal origin. She has muscular hypotonia, mental retardation, a cleft palate, mild hydrocephalus as a result of dilatation of both lateral ventricles, hyperextensible elbow joints, proximal radioulnar synostosis, clinodactyly of the fifth finger, valgus of the feet, and small hands and feet. In addition, she has a persistent pupillary membrane and congenital chorioretinal atrophy. The pathogenesis of pentasomy X is not clear at present, but it is thought to be caused by successive maternal non-dysjunctions.
- pentasomy X
- chromosomal abnormality
- mental retardation
- facial abnormalities
Statistics from Altmetric.com
Pentasomy X is a rare chromosomal abnormality that affects girls only, and is characterised by the presence of five X chromosomes instead of two. The first case of this chromosomal abnormality was reported in 1963.1 The condition is typically characterised by severe mental retardation, craniofacial malformation, short stature, and other physical abnormalities. The true incidence of pentasomy X is unknown at present, and the only known risk factor is female sex. We present a case of pentasomy X with maternal origin of the extra X chromosomes.
“The true incidence of pentasomy X is unknown at present, and the only known risk factor is female sex”
CASE REPORT
The patient was an 11 month old girl, the second child of healthy unrelated parents (mother aged 29, father 33). She was born by caesarean section during the 36th gestational week as a result of premature membrane rupture. Her birth weight was 2400 g (20–50th centile), length was 45.4 cm (25th centile), and head circumference was 34 cm (75–90th centile). Her sibling was healthy and normally developed. Cytogenetic analysis was requested on the third day after birth because the infant had a cleft palate with ventriculomegaly, and was performed on the patient’s cultured peripheral blood lymphocytes. All of the 20 cells analysed had an abnormal karyotype of 49,XXXXX (fig 1A). Both the parents and her sibling have a normal karyotype.

The results of a molecular and cytogenetic study in our case. (A) The karyotype of our patient showing 49,XXXXX (G banding; original magnification, ×1000). (B) Marker map, red arrows indicate informative markers and blue arrows non-informative markers. (C) The results of microsatellite analysis. The peaks of three informative X linked markers (DXS1001, DXS1047, and DXS8019) seen in the extra X chromosomes of our case are the same as those of her mother.
A molecular study using microsatellite markers (ABI Prism® linkage mapping set v2.5; Applied Biosystems, Foster City, California, USA) located on the X chromosome revealed that the peak area ratio of the mother and father in the patient’s informative markers was 3.99–4.59 to 1, indicating that her extra X chromosomes were maternal in origin (fig 1B). All analyses were performed on a 3100 ABI Prism genetic analyser™ (Applied Biosystems), according to the manufacturer’s instructions. Data were processed by GeneScan™ and Genotyper™ software (Applied Biosystems). The most typical features of this infant were muscular hypotonia, mental retardation, and mild hydrocephalus as a result of dilatation of both lateral ventricles. A magnetic resonance imaging scan of the brain was done to investigate the brain abnormalities, and showed dilatation of both lateral ventricles combined with mild leucomalacia in the periventricular area.
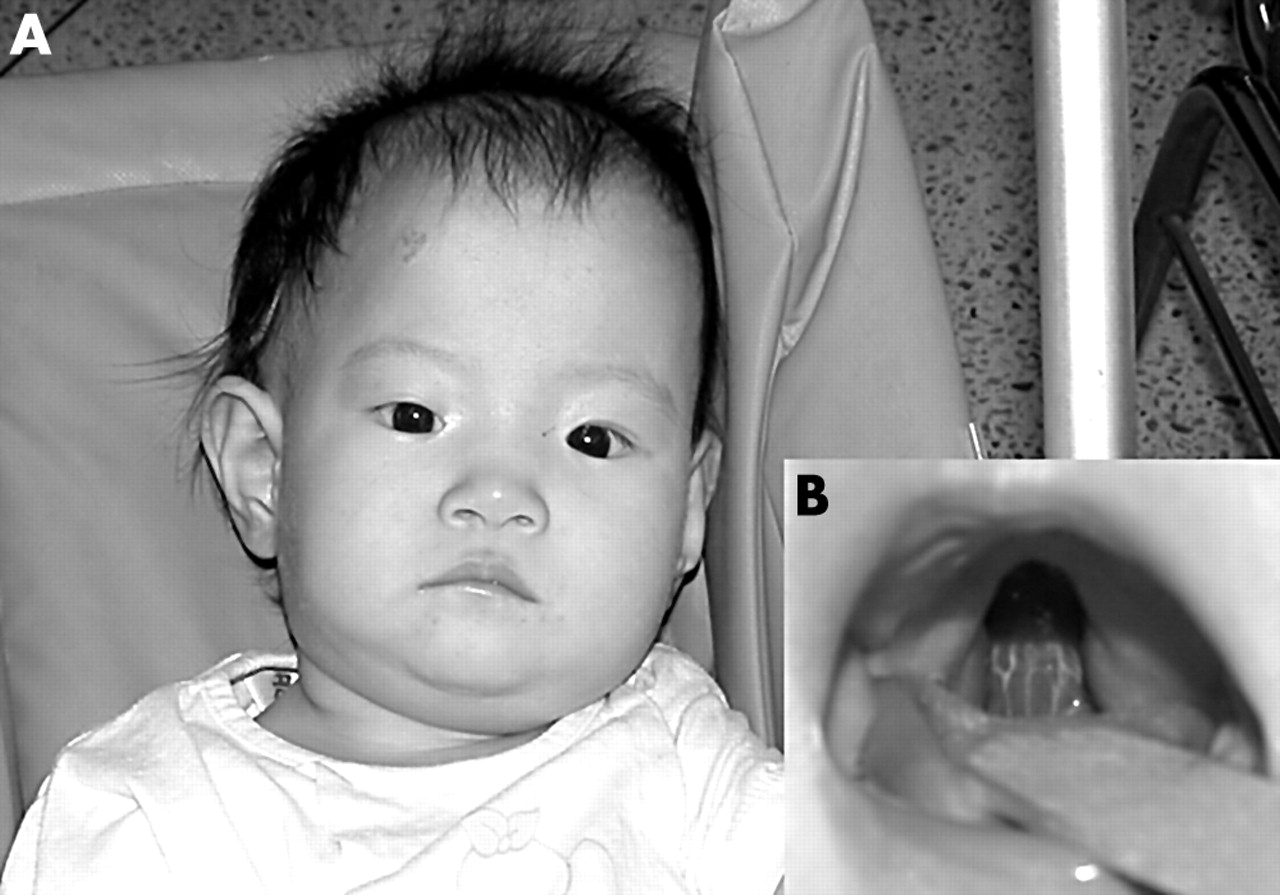
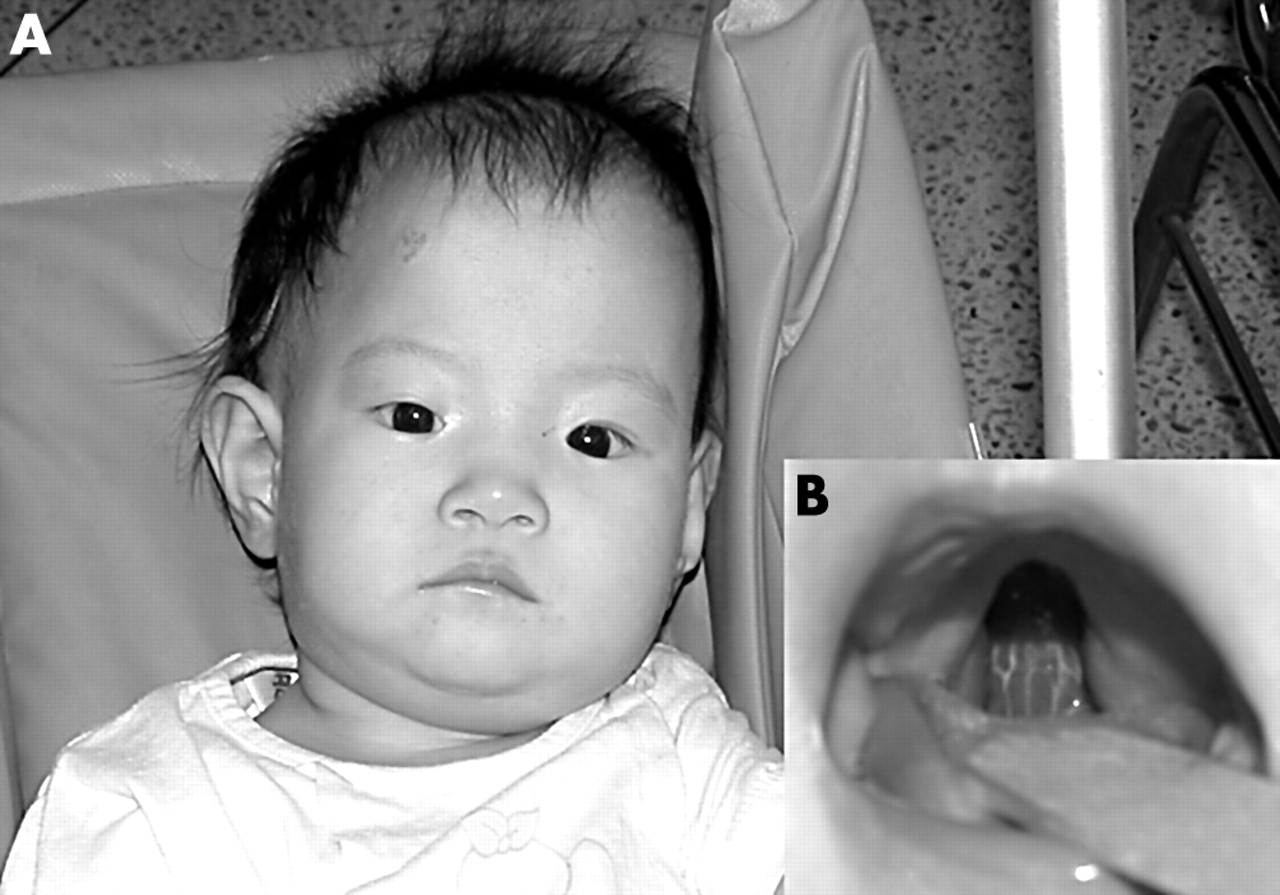
The general appearance of the patient showed the typical dysmorphism of pentasomy X (fig 2). Characteristic craniofacial features, such as hypertelorism, a flat broad nose, cleft palate, a persistent pupillary membrane, congenital chorioretinal atrophy, micrognathia, and malformed teeth, were found. She had a hyperextensible joint at the elbows, proximal radioulnar synostosis in the right forearm, clinodactyly of the fifth finger, small hands and feet, and valgus of the feet. A laboratory evaluation revealed a normal leucocyte count and a normal differential blood count. IgA (392 mg/litre), IgM (1280 mg/litre), and IgG2 (960 mg/litre) were low, although total IgG was normal (8720 mg/litre). There was no history of an increased incidence of recurrent infections.
{kind=link}
{kind=link}
(A) Photograph showing the facial appearance of the patient 11 months after birth. Characteristic facial features such as hypertelorism, a flat broad nose, and micrognathia are well portrayed. (B) Cleft in the soft palate of our patient.
DISCUSSION
Pentasomy X has several synonyms, namely: penta-X syndrome, chromosome X pentasomy, poly-X, and XXXXX syndrome. Numerical abnormalities of the sex chromosomes such as 47,XXX, 47,XXY, 47,XYY, and 45,X are relatively common, and occur in approximately 1 of 400 live births,2 but to our knowledge only about 30 cases of pentasomy X have been reported previously. This aneuploid state must arise as a result of a meiotic malfunction, either maternal or combined maternal and paternal in origin. The most likely mechanism is non-dysjunction of the maternal X chromosomes in both divisions of meiosis to produce an XXXX ovum, and this hypothesis has been supported by molecular analysis of X linked polymorphic markers.3 The microsatellite analysis used in our study showed that the extra X chromosomes seen in our patient were maternal in origin, probably as a result of successive maternal non-dysjunctions.
The parents of the patient were healthy and unrelated. The mother was 29 years old and the father was 33 years at the time of her birth, figures that are similar to previous reports.4,5 The age of the mother is one of the contributing factors in Down’s syndrome, but the influence of the mother’s age on the occurrence of penta-X syndrome has not been determined.
Because most patients with penta-X syndrome (including ours) have hypertelorism, epicanthus, and a mongoloid slant of palpebral fissures as facial anomalies, similar to the abnormalities seen in Down’s syndrome, occasional diagnostic problems have been reported.5 Thus, patients can be misdiagnosed as having Down’s syndrome, so that the correct diagnosis requires cytogenetic analysis.
The clinical manifestations seen in our case were consistent with those described previously in patients with pentasomy X.1,3–7 They included mental and developmental retardation, craniofacial anomalies, skeletal abnormalities, and cardiovascular anomalies. At first glance, our patient appeared to be normal, but she had multiple malformations and abnormal laboratory findings. The common features found in previously described patients and our patient were mental retardation, hypertelorism, mongoloid slant of palpebral fissures, a flat broad nose, malformed teeth, normal external genitalia, clinodactyly of the fifth finger, small hands and feet, and congenital heart disease. In contrast, features found frequently in previous patients and not in our patient included a short neck, a simian crease, and overlapping toes. A persistent pupillary membrane and congenital chorioretinal atrophy were unique to our patient. In addition, most of the other patients had varus of the feet, whereas our patient had valgus.
“Patients can be misdiagnosed as having Down’s syndrome, so that the correct diagnosis requires cytogenetic analysis”
The normal external genitalia seen in our patient have been reported in previous patients examined,1,4 although gonadal dysfunction has been seen in many cases, including a postmortem case.4,6,7 These findings suggest that despite the normal appearance of the external genitalia, there is an underlying gonadal dysfunction in patients with penta-X syndrome. Because our patient was an infant, her sexual development and bone maturation remain to be assessed.
Take home messages
-
We describe an 11 month old girl with features of pentasomy X: muscular hypotonia, mental retardation, a cleft palate, mild hydrocephalus as a result of dilatation of both lateral ventricles, hyperextensible elbow joints, proximal radioulnar synostosis, clinodactyly of the fifth finger, valgus of the feet, and small hands and feet
-
She also has a persistent pupillary membrane and congenital chorioretinal atrophy
-
Molecular and cytogenetic evaluation revealed that her karyotype was 49,XXXXX and her extra X chromosomes were of maternal origin
-
Pentasomy X may be caused by successive maternal non-dysjunctions
Immunoglobulin values in our case were similar to those reported previously in a 49,XXXXX female patient,3 including reduced serum IgA and IgG2 and normal total IgG. Our patient’s serum IgA and IgM values were inappropriately low compared with age related normal values. However, no history of a greatly increased incidence of recurrent infections was found in our patient, whereas Boeck et al described their patient as having a lifelong history of eczema, recurrent pneumonia, and staphylococcal abscess.3
According to a review of pentasomy X, mental retardation was seen in all of the 22 cases reported previously.8 Delayed psychomotor development from early infancy, as seen in our patient, has also been described in this syndrome.