Article Text

Abstract
Congenital heart disease (CHD) is the most common of congenital cardiovascular malformations associated with birth defects, and it results in significant morbidity and mortality worldwide. The classification of CHD is still elusive owing to the complex pathogenesis of CHD. Advances in molecular medicine have revealed the genetic basis of some heart anomalies. Genes associated with CHD might be modulated by various epigenetic factors. Thus, the genetic and epigenetic factors are gradually accepted as important triggers in the pathogenesis of CHD. However, few literatures have comprehensively elaborated the genetic and epigenetic mechanisms of CHD. This review focuses on the etiology of CHD from genetics and epigenetics to discuss the role of these factors in the development of CHD. The interactions between genetic and epigenetic in the pathogenesis of CHD are also elaborated. Chromosome abnormalities and gene mutations in genetics, and DNA methylations, histone modifications and on-coding RNAs in epigenetics are summarized in detail. We hope the summative knowledge of these etiologies may be useful for improved diagnosis and further elucidation of CHD so that morbidity and mortality of children with CHD can be reduced in the near future.
- cardiovascular system
- child health
- genetics
- pediatrics
Data availability statement
Data are available in a public, open access repository.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Congenital heart disease (CHD) is a group of disorders attributed by abnormalities in fetal heart and large vessels that lead to actual or potential impairment of cardiac function in infants. CHD is the most common type of congenital defect worldwide and is the most common and the most life-threatening class of birth defects in infants, affecting approximately 1% live births annually worldwide.1 Epidemiological investigations indicate that the overall incidence of CHD varies across countries and continents, and the prevalence of CHD in Asia is higher than that in North America.2 Despite benefits from the remarkable progress in therapeutic strategies of surgery and catheter intervention, CHD is still the principal source of mortality in infants. However, owing to medical, surgical and technological evolutions during the past decades, more than 90% of CHD infants now survive to adulthood.3 Improvement in surgical intervention techniques and perioperative care has dramatically changed the management of these populations with CHD. However, CHD is still a bothersome question owing to its undesirable outcomes and expensive healthcare costs, which bring substantial physiological, emotional and socioeconomic challenges to patients, families and society.
According to the final anatomical and pathophysiological complexities, CHD can be classified as mild, moderate or severe. Detailed classification is shown in box 1.4 The prognosis, morbidity and mortality vary with the severity of the anomalies. Despite the rapid advances in medical care and detection technology, the etiology of most CHD remains poorly understood. It is therefore imperative to improve our understanding of the disease mechanisms to reduce the frequent occurrence of CHD. During the past decades a consensus has emerged that both genetic (eg, chromosomal abnormalities, smaller copy number variants and point mutations) and environmental (extrinsic factors, such as teratogen exposure and nutrient deficiencies; intrinsic factors, including maternal disease, illness and viral infection)5 factors are related to the occurrence of CHD. Progress in molecular genetic diagnosis has provided a valuable opportunity to investigate the genetic factors of CHD. Furthermore, a multitude of animal models (eg, mouse, zebrafish, frog and fruit fly) have witnessed the significant effects of genetic etiology of CHD. These in vivo studies on animal models, in turn, have resulted in the identification of numerous structural genes, transcriptional regulators and signaling molecules that are critical for normal cardiac morphogenesis.6
Classification of congenital heart disease (the copyright can be viewed in the online supplemental file)
Mild:
Isolated congenital aortic valve disease and bicuspid aortic disease.
Isolated congenital mitral valve disease (except parachute valve and cleft leaflet).
Mild isolated PS (infundibular, valvular and supravalvular).
Isolated small ASD, VSD or PDA.
Repaired secundum ASD, sinus venosus defect, VSD or PDA without residuae or sequellae, such as chamber enlargement, ventricular dysfunction or elevated pulmonary artery pressure.
Moderate (repaired or unrepaired where not specified; alphabetical order):
Anomalous pulmonary venous connection (partial or total).
Anomalous coronary artery arising from the PA.
Anomalous coronary artery arising from the opposite sinus.
AS subvalvular or supravalvular.
AVSD, partial or complete, including primum ASD (excluding pulmonary vascular disease).
Coarctation of the aorta.
Double-chambered right ventricle.
Ebstein anomaly.
Marfan syndrome and related HTAD and Turner syndrome.
PDA, moderate or large unrepaired (excluding pulmonary vascular disease).
PPS.
PS (infundibular, valvular and supravalvular), moderate or severe.
Sinus of Valsalva aneurysm/fistula.
Sinus venosus defect.
TOF repaired.
Transposition of the great arteries after arterial switch operation.
VSD with associated abnormalities (excluding pulmonary vascular disease) and/or moderate or greater shunt.
Severe (repaired or unrepaired where not specified; alphabetical order):
Any CHD (repaired or unrepaired) associated with pulmonary vascular disease (including Eisenmenger syndrome).
Any cyanotic CHD (unoperated or palliated).
Double-outlet ventricle.
Fontan circulation.
IAA.
Pulmonary atresia (all forms).
Transposition of the great arteries (except for patients with arterial switch operation).
Univentricular heart (including double inlet left/right ventricle, tricuspid/mitral atresia, hypoplastic left heart syndrome and any other anatomic abnormalitywith a functionally single ventricle).
Truncus arteriosus.
Other complex abnormalities of atrioventricular and ventriculoarterial connection (ie, crisscross heart, heterotaxy syndromes and ventricular inversion).
AS, aortic stenosis; ASD, atrial septal defect; AVSD, atrioventricular septal defect; CHD, congenital heart disease; HTAD, heritable thoracic aortic disease; IAA, interrupted aortic arch; PA, pulmonary artery; PDA, patent ductus arteriosus; PPS, peripheral pulmonary stenosis; PS, pulmonary stenosis; TOF, tetralogy of Fallot ; VSD, ventricular septal defect.
To our knowledge, although numerous literatures have discussed the genetic mechanisms of CHD, few have comprehensively elaborated the genetic and epigenetic mechanisms of CHD. In this review, we focus on CHD origin from the etiology of genetics and epigenetics. Chromosomal abnormalities and gene mutations in genetics, and DNA methylations, histone modifications and on-coding RNAs in epigenetics are summarized in detail. Moreover, we expect that rapidly emerging data could provide a further understanding of genetics and epigenetics in the development of CHD and also a basis for further exploring the early diagnosis and individualized therapy of CHD.
Chromosome abnormalities underlying CHD
Chromosome abnormalities refer to abnormal chromosome numbers and structural aberrations including aneuploidies and copy number variations (CNVs), respectively. Conventional chromosome anomalies associated with CHD were identified half a century ago. A study by Pierpont et al7 suggested that ~30% of children with a chromosomal abnormality would suffer from CHD. In the following section, we summarize the involvement of chromosomal aneuploidies and CNVs in CHD in detail.
Aneuploidy in CHD
Chromosomal aneuploidy is the earliest recognized genetic cause of CHD and accounts for a great proportion of CHD (table 1). Approximately 50% of individuals born with trisomy 21 have the phenotypes of CHD, ranging from atrial septal defect (ASD)/ventricular septal defect (VSD) to atrioventricular canal lesions.1 2 The prevalence of CHD in newborns with trisomy 13 and trisomy 18 increases to 80%, and the major phenotypes of CHD are heterotaxy, laterality and septal defects.8 9 CHD is observed in approximately 33% of females with Turner syndrome or monosomy X, and the cardiac malformations are usually diagnosed as VSD, coarctation of aorta (CoA), bicuspid aortic valve and hypoplastic left heart.1 Although abnormal X chromosome numbers are rare, they also could result in CHD. For example, males with Klinefelter syndrome or 47, XXY have a 50% chance of CHD with the phenotypes of patent ductus arteriosus (PDA) and ASD.7 Moreover, sporadic 49, XXXXX cases with the phenotypes of ASD and vascular malformations have also been reported.10 At present, chromosomal G-banded karyotype analysis has been applied to detect the anomalies of chromosomes in spite of its limitation for the restrictive base resolution in exploring the tiny abnormalities of chromosomes.
Common congenital heart disease resulting from chromosomal abnormalities
CNVs in CHD
Conventional chromosomal microscopy in clinic could only detect the alterations of structure, numbers of chromosomes and abnormalities of large fragments, causing incomplete diagnosis of CHD. Numerous detection technologies include fluorescent in situ hybridization and multiplex ligation-dependent amplification. Chromosomal microarrays have been applied to explore the submicroscopic chromosomal anomalies and to elucidate the pathogenic mechanisms of CHD, which may become a better approach for diagnosis.
CNVs refer to structural aberrations consisting of deletions or duplications, which are too small to be detected by routine karyotype analysis. A few CNVs can alter one or more contiguous genes and then inappropriately affect their expression, leading to the development of CHD.11 12 CNVs can occur de novo in sporadic cases, or they can be inherited familiarly causing complex congenital heart malformations. However, the underlying mechanisms of CNVs in CHD still need to be elucidated. One of the most common CNVs syndromes in CHD, 22q11.2 deletion syndrome (DiGeorge syndrome or velocardiofacial syndrome), is caused by a microscopic deletion on chromosome 22q11.2. Variable phenotypes, such as craniofacial abnormalities, neurocognitive disabilities, palate abnormalities, hypocalcemia and immunodeficiencies, can be observed in this syndrome. The main cardiac malformations of this syndrome contain VSD, arch abnormalities and tetralogy of Fallot (TOF).13 To date, more than 30 gene deletions have been identified in the 22q11.2 locus by sequencing. Deletions in the T-box transcription factor TBX1 account for the major molecular basis of these cardiac malformations.14 Further work revealed that distal 22q11.2 deletion presented atypical clinical features of DiGeorge syndrome; therefore, this deletion was proposed as one of the etiologies of CHD.15 Williams-Beuren syndrome, which is characterized by supravalvar aortic stenosis (SVAS), peripheral pulmononic stenosis, coronary artery stenosis, pulmonary artery sling, developmental delays, typical elfin facies, infantile hypercalcemia and cognitive disability, has been reportedly caused by microdeletion of over 25 genes in the 7q11.23 region.16 It has been known that cardiovascular abnormalities, such as SVAS, can be induced by haploinsufficiency of the elastin gene (ELN).17
Some CNVs encompass previously identified CHD genes, or genes known to be implicated in heart development. For example, 8 p deletion syndrome and mutations in GATA4, a cardiac transcription factor, are reported to be associated with CHD.18 The 8p23.1 delete region that overlaps with the locus of GATA4 could elucidate this causal outcome. However, Kumar et al reported an interesting case of 8p23.3p23.1 deletion and 8p23.1p11.1 interstitial duplication syndrome that a male toddler with global developmental delay, dysmorphic facies, seizures and large doubly committed VSD occurred without the GATA4 gene involvement.19 Duplications at the 8p23.1 locus have also been identified in CHD, including ASD, VSD and TOF.20 21 Other studies have confirmed that the CNVs at chromosome 11q23 were associated with Jacobsen syndrome.22 23 Moreover, several CNVs have been identified from larger cohorts of patients with CHD, including 1q21.1,24–26 4p16.3,27 4q22.1,26 28 9q34.326 29 and 15q11.2.28 All of these CNVs mentioned above are detailed in table 1 for their relationship to CHD in human.
Gene mutations underlying CHD
Numerous mutations are implicated in the development of CHD (table 2). Some mutations are identified in pedigrees of CHD, while others are initially observed in sporadic cases of CHD.2 Currently, mutations in more than 50 genes have been found to be associated with CHD by the application of high-throughput sequencing of whole-genome and whole-exome, and many of these affected genes have been confirmed to be involved in transcriptional regulation, signal transduction and cardiac development.
Common congenital heart disease resulting from single gene defects
Mutations of genes encoding transcription factors in CHD
Heart development is regulated by several transcriptional circuits that are members of a core group of transcription factors, including NKX2.5, GATA4 and TBX5.30 31 Therefore, transcription factors have been considered as the prime inducer of CHD. NKX2.5 is the earliest known marker of myocardial progenitor cells in all species.32 The mechanisms of NKX2.5 regulation and its interaction with other transcription factors in early cardiac development have been studied extensively. It has been found that mutations in NKX2.5 could result in variable types of CHD, including ASD, VSD, TOF, hypoplastic left heart syndrome, CoA, transposition of the great arteries, double outlet right ventricle (DORV), interrupted aortic arch and cardiac outflow tract defects.33 Furthermore, NKX2.5 mutations are the most common cause of ASD in individuals with defects of conduction system.34 35
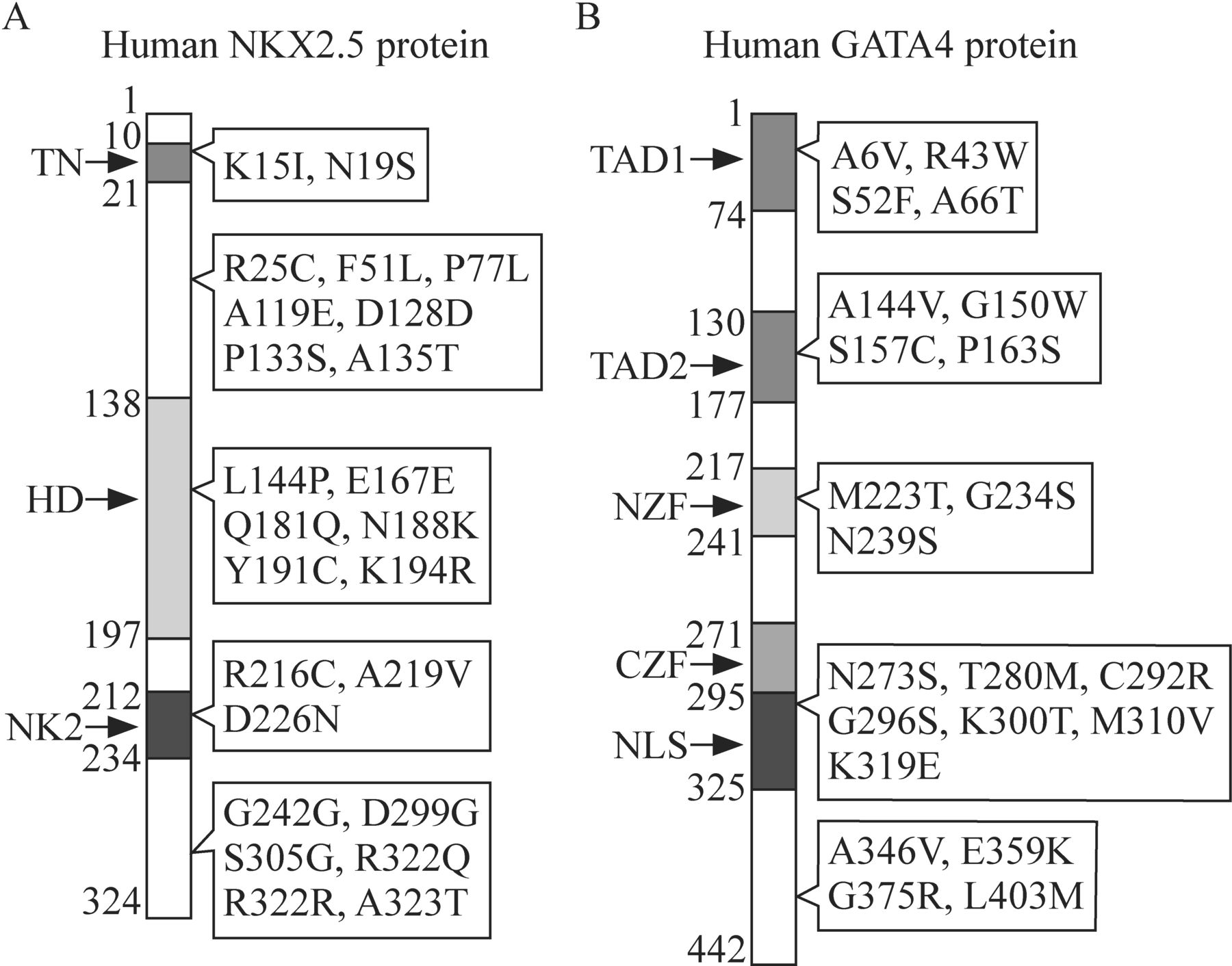
However, some individuals with CHD caused by the mutations of NKX2.5 can manifest an individual phenotype of ASD and/or conduction defects.36 To date, approximately 80 different mutations have been identified in NKX2.5, including missense mutations [eg, c.44A>T (p.K15I), c.232A>G (p.N19S), c.673C>A (p.N188K) and c.1089A>G (p.S305G)], synonymous mutations [eg, c.543G>A (p.Q181Q), c.677A>G (p.E167E), c.902C>G (p.G242G) and c.1142A>G (p.R322R)] and nonsense mutations [eg, c.1149T>C (stop→Gln)].33 The location of some of these mutations is depicted in figure 1A. Holt-Oram syndrome, which is characterized by CHD (eg, ASD, VSD and atrioventricular conduction system disease) and upper limb malformations, could be caused by the loss-of-function mutations in TBX5, which is notably expressed in the upper limbs and heart.37 38 Nonsense or frameshift mutations of TBX5 may be responsible for this syndrome.12 Functional deficiency of the conserved DNA-binding motif in the transcription factor encoded by TBX5 may be the etiology of Holt-Oram syndrome.39

Spectrum of some significant mutant sites of (A) NKX2.5 and (B) GATA4 observed in the occurrence of congenital heart defects. CZF, C-terminal zinc finger domain; HD, homeodomain; NK2, NK-2-specific domain; NLS, nuclear localization signal; NZF, N-terminal zinc finger domain; TAD1, transcription activation domain 1; TAD2, transcription activation domain 2; TN, tin-man domain/transcriptional repression domain.
GATA4, encoding one of the GATA zinc-finger transcription factors, is a deeply studied gene and is essential for cardiogenesis. Mutations in GATA4 are reported to be implicated in cardiac septal defects.31 Over 100 mutations in GATA4 coding region have been identified in patients with CHD, and more mutations will be identified with intensive research.40 Among these mutations, 11 sites (two synonymous mutations, seven missense mutations and two frameshift mutations) have been studied in familial cases, which highlight the significance of these sites in the development of CHD (figure 1B). Multiple mutations identified from other transcription factors, including NKX2.6,41 GATA5,42–44 GATA6,45 TFAP2β,46 TBX1,47TBX2048 49 and ZIC3,50 have also been reported to be associated with the incidence of CHD.
Mutations of genes encoding signal proteins in CHD
Signaling pathways involved in the occurrence of CHD are widely studied. Genes involved in these different signaling pathways can converge into a large and sophisticated regulatory network that plays an important role in cardiac development and pathogenesis of CHD. Recent studies have also suggested the potential contributions of vascular endothelial growth factor-A (VEGF-A), Notch signaling, Wnt signaling, transforming growth factor-β (TGF-β), bone morphogenic protein (BMP) signaling and cpathway to the occurrence of CHD.51–57 In these pathways, some are essential for the formation of cardiac septum, valves and the construction of cardiac outflow tracts, while others are associated with the asymmetric development of the heart. Gene mutations in renin–angiotensin system mitogen-activated protein kinase (RAS-MAPK) signal transduction pathway can lead to Noonan syndrome with the typical phenotype of pulmonary valve stenosis and hypertrophic cardiomyopathy.58–61
Six missense variants (COL6A1, COL6A2, CRELD1, FBLN2, FRZB and GATA5), acting in the VEGF-A pathway, were found to be damaged in individuals with complete atrioventricular septal defect (AVSD),51 suggesting that rare variants in the VEGF-A pathway might play a role in the development of AVSD. In addition, mutations in VEGF-A have been reported to be associated with congenital left ventricular outflow tract obstruction.62 Notch signaling is a highly conserved pathway involved in developmental process of heart. JAG1 encodes a ligand in the Notch signaling pathway, which leads to localization of Notch to the nucleus and downstream activation of target genes. Mutations in JAG1 have been found in over 90% cases of Alagille syndrome.63 64 Some cases (~2%) that have mutations in NOTCH2, a NOTCH receptor gene, are also correlated with Alagille syndrome.65 However, variants of NOTCH1 that belongs to the Notch signaling pathway have been identified to be associated with Adams-Oliver syndrome.15 Mind bomb 1 (Mib1) is a vital protein that promotes ubiquitination, endocytosis and subsequent activation of Notch ligands to activate the Notch signaling pathway. Mutations in MIB1 have been identified to be associated with cardiac deformity such as ASD, AVSD and VSD, through a lower level of JAG1 ubiquitination and Notch signaling induction.66 In addition, mutations in other genes of Notch signaling pathway include MAML1,67 DLL468 and GALNT1169 are also involved in CHD.
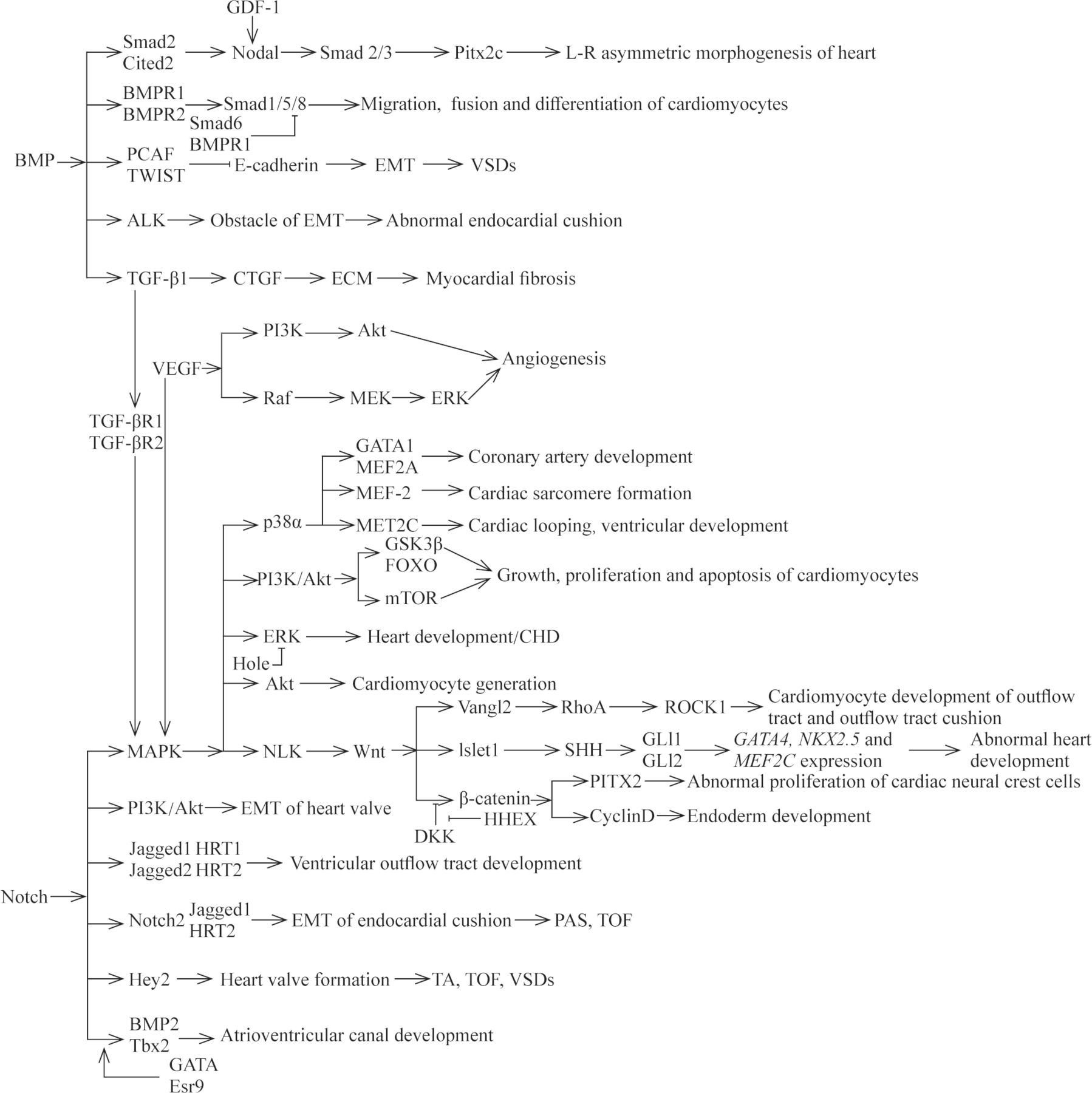
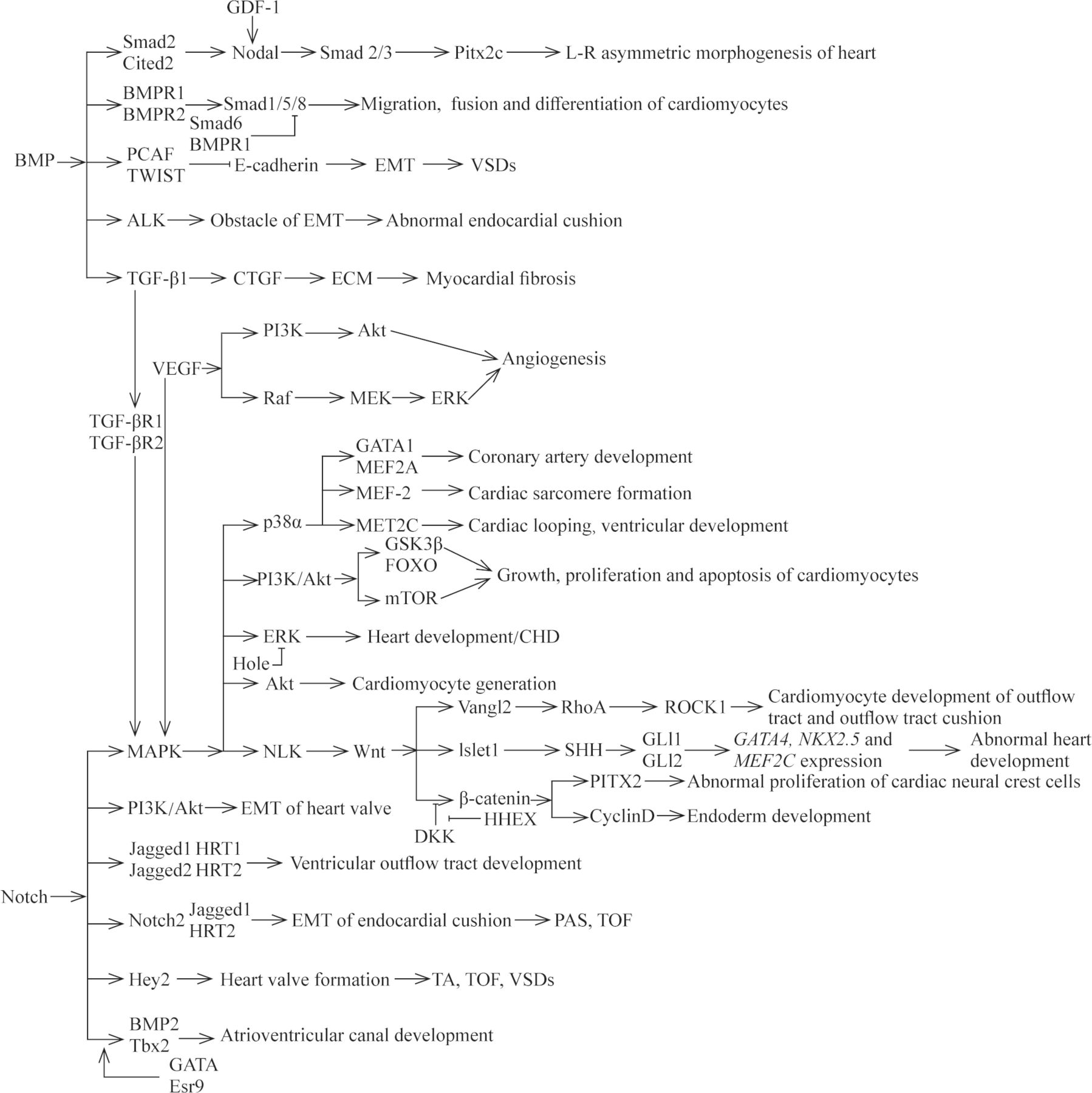
AXIN2 is involved in the regulation network of cardiac valve formation and elongation, and its expression product is a negative regulator of Wnt/β-catenin signaling pathway.70 71 It has been found that mutations in AXIN2 can result in CHD with the phenotype of congenital valve defect.72 HHEX, a member of the Homeobox gene family, is an important cardiac determinant and controls the early differentiation, migration and development of cardiomyocytes.73 Foley et al74 reported that mutations in HHEX could lead to a phenotype of abnormal developmental endogenous cardiac or ectopic heart, which was similar to the antagonistic effect of Dickkopf-1 to Wnt signaling pathway. Aberrant Wnt signaling pathways implicated in CHD have been summarized in a previous excellent literature.75 We describe the aberrant expression of genes associated with CHD within the Wnt signaling pathways in figure 2. TGF-β signaling pathway has important role in the development and remodeling of cardiovascular system. Aberrant TGF-β signaling pathway is involved in the pathogenesis of several human cardiovascular diseases through the epithelial-to-mesenchymal transition (EMT) of resident fibroblasts, circulating progenitors, pericytes, epithelial cells and/or the endothelial-mesenchymal transdifferentiation of endothelial cells.76 77 For example, mutations in TGF-β1, one major subtype of TGF-β family, have been reported to be associated with CHD in pediatric patients.78 In general, BMP synergizes with TGF-β signaling to activate the downstream genes, such as Smad1, Smad5 or Smad8, with the changed transcription of target genes. Mutations in the genes that encode transducers of the TGF-β and BMP signaling pathway have been identified in the pathogenesis of cardiovascular diseases, such as Marfan syndrome and Loeys-Dietz syndrome.55 79 Nodal signaling pathway is involved in the left–right patterning and development of the heart and in abnormal gene products throughout the pathway that are clearly associated with CHD. Roessler et al56 previously demonstrated that reduced nodal signaling strength via mutation of FOXH1 was linked to human heart defects. SHH, one morphogen of hedgehog (HH) family proteins, is involved in a remarkably wide variety of process, including cardiovascular development. Previous evidence demonstrated the mutations in genes of HH signaling were implicated in the occurrence of CHD with a phenotype of ASD, VSD or AVSD, and the responsible mutated genes include SHH, Gli3 and MKS1.80–83 Noonan syndrome, one of the most common genetic syndromes of CHD, is caused by mutations in genes of the RAS-MAPK pathway.84 At present, several genes have been verified to be responsible for the development of Noonan syndrome and other disorders. Mutations in genes, encoding molecules implicated in the RAS-MAPK signaling pathway, account for approximately 90% of affected CHD cases. These genes include: PTPN11, SOS1, KRAS, RAF1, BRAF, SHOC2, NRAS, HRAS, CBL, MEK1, MEK2, PPP1CB, RIT1 and SOS2.15 58 61 84
{kind=link}
{kind=link}
The relationships of different signaling pathways and some responsible mutated genes within these pathways in cardiac development and CHD. CHD, congenital heart disease; ECM, extracellular matrix; EMT, epithelial-mesenchymal transition; PAS, pulmonary artery stenosis; TA, tricuspid atresia; TOF, tetralogy of Fallot; VSD, ventricular septal defect.
Currently, most studies have only stayed on the basic exploration of a single gene, which cannot fully explain the pathogenesis of CHD. The interactions of various signaling pathways involved in heart development are still incompletely understood. Some responsible mutated genes have been proposed to clarify the interactions of the signaling pathways implicated in CHD (figure 2). More researches are needed to help elaborate the pathogenic mechanisms of CHD for the intricate network of signaling pathway.
Mutations of genes encoding cardiac structural proteins in CHD
Several studies use targeted whole-genome sequencing to investigate the genes that encode cardiac structural proteins to elucidate the monogenic cause of CHD. Some rare missense mutations or premature termination mutations in myosin heavy chain 6 (MYH6), a marker gene of myocardial cell, could result in ASD.85 86 However, mutations in MYH7 have been confirmed in bilobal aortic valve.87 CHD with apical hypertrophic cardiomyopathy, left ventricular non-compaction and ASD could be caused by the mutations in ACTC, which is a cardiac actin gene and is essential for cardiac contraction.88 89 MYH11, encoding smooth muscle myosin heavy chain, is another gene that has been reported to be involved in CHD with the phenotype of dominant thoracic aortic aneurysm.90 91 Moreover, loss-of-function mutations in the elastin gene (ELN) could result in CHD with the presence of Williams-Beuren syndrome and non-syndromic SVAS.17
Epigenetic modifications underlying CHD
Epigenetics, which refers to the mechanisms of changed gene expression that are independent of DNA sequence, provides a new way to understand the pathogenesis of CHD. To date, three canonical mechanisms of epigenetics include DNA methylation, histone modification and non-coding RNAs. The increasing evidence suggests that the aberrant regulation of gene expression by epigenetics is a key factor in the development of cardiovascular diseases, which have attracted attention to focus on the role of epigenetics in CHD.
DNA methylation modifications in CHD
DNA methylation, the most widely studied epigenetic mechanism, refers to the formation of a methyl group (−CH3) in the 5′ carbon of cytosine (CpG islands), which induces an alteration of the structure of DNA. The methylation process is catalyzed by DNA methyltransferases (DNMTs), comprising DNMT1, DNMT3A and DNMT3B. The dysregulation of DNA methylation during different stages of development could lead to the transcriptional repression and functional inactivation of tissue-specific genes, resulting in increased risk for several diseases, including cardiac malformation. Alterations of DNA methylation, especially in CpG islands, which are close to core transcription factors and genes in signaling pathway, have been reported in patients with cardiac malformations.92
DNA methylation plays a critical role in the development of the heart. The expression of hyaluronan synthase 2 (Has2) is necessary for the formation of the heart valves, septa and epicardium. However, Has expression was found to be downregulated via DNA methylation in the heart at E14.5 embryos.93 Furthermore, gene knockout model indicated that expression of Has2 is downregulated via DNA methyltransferase 3B (DNMT3B), which was coexpressed with Has2 in the region of cardiac valve, suggesting that changes in DNA methylation might be involved in the regulatory function of Has2 enhancer. Aberrant methylation of CITED2 may play an important role in the development of VSD, ASD and TOF.94 Aberrant methylations of CITED2 could decrease its mRNA expression and be regarded as the prime cause of CHD.95 The presence of aberrant hypomethylation in the CpG region of BRG1 was found to exist in patients with ASD and interventricular septal defect.96 It has been shown that the promoter region of CX43 plays an essential role in the development of the heart outflow tract, and the aberrant hypomethylation of this enhancer region of CX43 could be considered as one of the etiologies of CHD.97 98
In another study, two encoding transcription factors, zinc-finger in cerebellum 3 and nuclear receptor subfamily 2 group F member 2, were found to be hypermethylated in monozygotic twins with DORV, suggesting that the differential methylation of these transcription factors could be regarded as a potential pathogenesis of the diseased twin.99 Hypermethylation of the promoter region of SCO2, a cytochrome oxidase, has been found in patients with TOF and VSD. The methylation of CpG islands located in the promoter of SCO2 result in reduced expression of SCO2, which may be the mechanism of the occurrence of these diseases.100 Further studies discovered that hypermethylation of CpG islands of NKX2.5, HAND1 and RXRα was also essential for CHD, including VSD and TOF.101 102 Multiple differential methylated genes have been identified from cardiomyocytes of newborn and adults, and adult failing hearts have revealed a highly dynamic of DNA methylation under specific developmental or pathological conditions. DNA methylation is tightly regulated during cardiac differentiation and maturation.103 Taken together, these findings highlight the importance of DNA methylation in cardiac morphogenesis and CHD formation.
Histone modifications in CHD
Histones, including H1, H2A, H2B, H3 and H4, and DNA constitute the nucleosome, which is the basic structural unit of chromatin in eukaryotic cells.104 Altering the histone-DNA contacts effectively via histone, post-translational modifications could loosen or tighten the chromatin architecture to control the availability of gene transcription or expression. Histone post-translational modifications could be modified by the catalysis of histone-modifying enzymes, such as histone methylases, demethylases, acetylases and deacetylases, ubiquitin enzymes and phosphorylases. Aberrant expression and mutation of the histone modifiers during the development of heart can influence the response of heart to pathological stresses.105 Moreover, increasing researches show that the interplay between these different cardiac transcription factors and histone modifiers plays a significant role in heart development. Numerous studies have shown that methylation and acetylation of histone is an emerging epigenetic mechanism for the regulation of gene transcription. Therefore, we focused only the mechanism of histone methylation and acetylation in CHD.
Methylation mainly occurs at the core histones H3 and H4, which is performed by the catalysis of histone methyltransferases. H3K4, H3K36 and H3K79 methylation leads to the transcriptional activation, whereas transcriptional repression can be induced by H3K9, H3K27 and H4K20 methylation.106 CHD associated with Wolf-Hirschhorn syndrome exists deletion of Wolf -Hirschhorn candidate protein 1 (Whsc1), which encodes the H3K36me3-specific methyltransferase. Nimura et al107 found that the pathogenic role of Whsc1 was associated with the transcriptional activation of Nkx2.5 in its target sites through the model of Whsc1-knockout murine. Another studies revealed that the interactions of JARID2 and SETDB1, which was an H3K9me3-specific methyltransferase, could elucidate the role of Jarid2 in the occurrence of VSD, DORV and impaired ventricular compaction induced by hypertrabeculation.108 109 TBX1, one member of T-box transcription factor family, was shown to interact with H3K4 and H3K27 via two domains of T-box to regulate gene expression, and aberrant expression could result in CHD including TOF, VSD and aortic arch interruption.110 111 A later study further identified the interactions of TBX1 and BAF chromatin remodeling complex regulate Wnt5a expression, and the insufficient expression of TBX1 or Wnt5a could result in the phenotypes of hypoplastic right heart.112 DPF3, an evolutionarily conserved protein, binds methylated and acetylated lysine residues of histone 3 and 4 to regulate gene expression. Dpf3 is expressed in the heart during development and has been found as significantly upregulated in the right ventricular myocardium of patients with TOF.113
Histone acetylation and deacetylation are always in a dynamic state, which is associated with transcriptional activation or transcriptional repression, gene silencing and cell cycle. Histone acetylation and deacetylation have been studied extensively in recent years.114 It has been shown that some histone acetylases, such as EP300, KAT2A and hNAT1, are associated with the development of the heart. Aberrant expression of these acetylases could lead to CHD with ASD, VSD, AVSD and valve dysplasia.115 116 Previous studies have revealed that histone deacetylases (HDACs) are also implicated in CHD. For example, HDAC3 regulates the cellular acetylation level affecting cardiogenesis, and the absence of HDAC3 could cause PDA and TOF.117 In addition, down-regulation of HDAC5 and HDAC9 simultaneously could result in CHD with VSD.118 119 Abnormal regulation of SIRT1 has been reported to be involved in ventricular hypoplasia.120 Park et al121 revealed that histone methyltransferase SMYD1 was associated with ventricular hypoplasia. Some other histone modifiers have been shown to be important for the development of the heart through numerous studies, such as G9a, Ezh2/PRC2, Baf60c/Brg1, Jmjd3, UTX and MLL2,122–130 and the aberrant modification of them could yield critical CHD phenotypes. The activity of cardiac transcription factor could be changed by histone modifiers, which means these modifiers could be strong candidates for CHD etiology and therapeutic targeting.
Non-coding RNA in CHD
Non-coding RNA is another type of epigenetic modification involved in the control of gene expression by post-transcriptional regulation. Among the various non-coding RNAs, microRNAs (miRNAs) and long non-coding RNAs (LncRNAs) are the two best studied groups. miRNAs are capable of regulating gene expression by interacting with mRNA transcript 3′ untranslated regions (UTRs) to repress translation, whereas LncRNAs are able to regulate transcription by directly interacting with chromatin remodeling complexes.131 Emerging evidence that indicates the impact of non-coding RNAs to cardiac development was previously unappreciated but is becoming valuable.
A number of miRNAs have recently been shown to function in the heart.132 It has been shown miR-1 is important in cardiac development and is regarded as the most relevant miRNA leading to CHD.133 134 Li et al found that expression of miRNA-1 decreased in patient with VSD.135 Molecularly, miRNA-1 binds to its target gene GJA1 and SOX9, regulating the formation of cardiac valves and septa, and therefore miRNA-1 dysregulation could result in CHD in human.135 Further studies have demonstrated that myocyte enhancer factor 2 (Mef2) could upregulate the expression of miR-1, which suppress the cardiac transcription factor Hand2 and HDAC translation.133 136 Hand2 is known as a target of miRNA-1 and is involved in the growth of the embryonic heart; thus, Hand2 could be associated with CHD.133 Overexpression of miRNA-27b could repress the expression of Mef2c and affect the development of myocardial, leading to cardiac hypertrophy.137 A recent study has confirmed that differentially expressed frataxin (FXN) regulates the development of CHD and that differential expression of FXN is under the control of miRNA-145.138 Further investigation of this study demonstrated that overexpression of miRNA-145 could regulate apoptosis and mitochondrial function by repressing the expression of FXN, leading to the development of CHD.138 In recent years, increasing evidence has confirmed that alterations of miRNAs expression are associated with human cardiovascular diseases, including CHD (reviewed by Nagy).139 Furthermore, miRNAs are extensively studied as clinical biomarkers for their stability in blood, urine and other biological fluids and their ability to evade RNA degrading enzymes. The researchers demonstrated that miRNAs in maternal serum could be used as candidate biomarkers for prenatal detection of fetal CHD in early pregnancy.140 141 These authors identified miR-19b and miR-29c significantly upregulated in patients with VSD, while miR-19, miR-22, miR-29c and miR-375 upregulated in patients with TOF. In recent years, increasing new miRNAs have been found to be associated with CHD, suggesting a potential value of miRNAs as diagnostic markers in human cardiovascular diseases.
LncRNA regulates cell growth, differentiation, cell proliferation and apoptosis by controlling gene expression. To date, little information of the functional role of lncRNAs in CHD has been reported, and only scant evidence has demonstrated the involvement of lncRNAs in CHD, particularly on VSD and TOF. Jiang et al142 reported the increased expression of SNHG6 in fetal cardiac tissues of VSD patients and suggested SNHG6 might be involved in VSD by the mechanistic link between SNHG6 upregulation, miR-100 downregulation, Wnt/β-catenin activation and the formation of VSD. The author also identified that expression of HOTAIR was increased in right atrial biopsies of CHD patients with ASD and VSD and postulated HOTAIR as a biomarker for CHD. Another study identified a polymorphism of MALAT1 associated with ASD and VSD.143 LncRNA TUC40 has been reported to reduce the expression of PBX1 and to affect the differentiation of cardiomyocytes, which may be a potential pathological etiology of VSD.144 In addition to the importance of LncRNAs in cardiac septal defects, the role of LncRNAs in the development of cyanotic heart disease such as TOF has been reported. Wang et al145 indentified that high expression of HA117 was associated with adverse outcomes in TOF patients, although the mechanism of HA117 in TOF remained unclear. In a recent study by Gu et al,146 the circulating plasma LncRNAs have been implicated in cardiovascular diseases for its potential as new biomarkers of diagnostic and prognostic in clinical treatments. The aberrant expression of the following LncRNAs: ENST00000436681, ENST00000422826, AA584040, AA709223 and BX478947 are associated with CHD. These specific LncRNAs identified from the plasma of pregnant women with typical fetal CHD may play an important role in the development and prenatal diagnosis of fetal CHD.
Conclusions
Normal development of the heart is a complex process involving many regulatory factors, including genetics and epigenetics. Cardiac malformations are congenital developmental disorders that can be induced by the dysregulations of genetic and epigenetic factors. Abundant seminal studies have found that conventional genetic factors could not elucidate the pathogenesis of CHD alone. Epigenetic regulation is also an important aspect of normal cardiac development as well as of defective function in disease situations such as CHD. The interactions at multiple levels provide insights of the combinatorial regulation into the morphogenesis of heart and suggest they can partially compensate each other’s function. Phenotypic heterogeneity and incomplete penetrance of CHD complicate our understanding of the interactions between genetics and epigenetics in CHD. Future studies to focus on elucidating the epigenetic signals of genes associated with cardiac development pathways could throw light on the genetic and epigenetic mechanisms in the development of CHD.
With the great progress in basic researches carried out in model systems (eg, in vivo: animal models; in vitro: cell and tissue engineered models) and with the improving limits of detection, the study of CHD has entered a new era for clearer understanding of its etiology. The wide application of these new detection technologies would provide an effective method for the prevention, diagnosis and treatment of CHD, opening up new avenues of individualized care for patients with CHD.
Supplemental material
Data availability statement
Data are available in a public, open access repository.
Ethics statements
Ethics approval
Not required for this review article.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
YW and XJ contributed equally.
Contributors YW and XJ contributed to conceptualization and data curation. YW, XJ, YZ and JZ performed writing and revising the original draft. RY was responsible for conceptualization and conducted all procedures, including data curation, writing the original draft and revised manuscript. All authors read and approved the final manuscript.
Funding This work was supported by the National Key R&D Program of China (grant number 2018YFC1002700) and the National Key R&D Program of China (grant number 2018YFC1002703).
Disclaimer The funder of the work had no role in: study design, or collection, analysis, and interpretation of data, or writing the manuscript or decision to submit.
Competing interests The authors declare that they have no conflicts of interest to report regarding the present study.
Provenance and peer review Not commissioned; externally peer reviewed.